Here’s a game I’m playing with my internet friends in 2026.
This is designed to be multiplayer and played across different regions. It will definitely work better if a bunch of people are playing in the same area based on the same list, but since we’re not, whatever, it’ll probably be hella unbalanced in unexpected ways. Note that the real prize is the guys we found along the way.
The game is developed using iNaturalist as a platform. You can probably use a field guide or a platform like eBird too.
PHILOSOPHY
First, I watched a bunch of Jet Lag: The Game, and talked with my friends about competitive game design using real-world environments. Then we watched the 2025 indie documentary Listers: A Look Into Extreme Birdwatching, which is amazing, and free. It’s about two dudes who are vaguely aware of birds and decide to do a “Big Year”, a birdwatching competition of who can see the most bird species in the lower 48 states. And I thought wow, I want to do something like that.
Nature is cool and I want to learn more about it. But I’m not personally that worked up about birds. Also, my friends and I all live in different places, many on shoestring budgets. So we were going to need something else.
This is my attempt at that: SPECIESQUEST. It’s a deeply experimental, distributed, competitive species identification game. It’s very choose-your-own-adventure – designed so that players can choose a goal that seems reasonable to them and then play against each other, making bits of progress over the course of a year (or whatever your chosen play period is). Lots of it relies on the honor system. It might be totally broken as is and I’m missing obvious bits of game design as well, so we’ll call this V1.
SETUP
There are two suggested ways to play: Local % and Total Species.
In Local %, you’ll try to find as many species (within whatever category or categories you like) as possible, that exist within a specific region you spend time in. I suggest this if you want to get to know a place better.
In Total Species, your goal is to maximize the # of species you observe and record on iNaturalist, potentially within a specific category of interest (herbaceous plants, fish, whatever). I tentatively recommend this if you travel and want to play while in other places, or want to be maximally competitive, or find the checklist-generation process for Local % too confusing.
(It’s pretty easy to switch between them later in the year if you feel like it.)
Local %
To play Local %, you’ll come up with a checklist of all the species known to exist for your region. Only observations within that region count.
The Checklist
First, come up with your CHECKLIST.
You can find a FIELD GUIDE to your area and use everything – perhaps in some given category – as your LIST.
But this is the modern age, and in iNaturalist, here’s how I did it:
Click “Explore” to look at existing observations.
Choose a region. I chose the county I live in. The bigger it is, the more you might have to travel to find candidates. I believe there are ways to create your own boundaries too in iNaturalist, but I’m not certain.
Go to “Filters”. Narrow down the phylum/candidates you want.
E.g. to get to “lichen”, I clicked the “fungi including lichens” box, then I added “lichen” in the description.
I strongly recommend specifying “wild” observations. See the Wild vs Domestic section under Everyone should think about scoring further down.
Select the grade of observations you want to include on your list. “Research grade” will return sightings that very clearly identify the species, IE of species that are really likely to actually be in your area.
Play with these until you have a goal that seems reasonable to you.
Once you have a list you’re happy with, save it. This is your CHECKLIST.
Search your area and identify species over the course of the year.
If you’re in your area and observe a species that’s NOT on your checklist, e.g. there is no iNaturalist existing info about it in that area, you can still count it. You DO have to identify it. That means it is possible to get a score of over 100%.
You can play in multiple categories at once. Just add them up to score. (e.g. if your region has 10 birds and 25 trees, your final score will be out of 35.)
Total Species
Go out and identify as many different species as possible.
Optional: In advance, choose a category to play within. If you’re really interested in birds, this might help you avoid some failure mode like “I was hoping to get more into birdwatching but I keep racking up all these plant identifications because it’s so much easier to find them and they stay still.” You’re playing for the Total Bird Species crown.
Roll your own?
Feel free to choose some other species-counting scoring criteria. Your SPECIESQUEST is your own.
Everyone should think about scoring in advance
Which observations count?
Think about this now. “Clear enough to identify the species” is the general heuristic.
I guess in the birding scene the proof of existence is photos and calls. If you are playing with lichens, probably the call will not be relevant.
“Clear observations on iNaturalist” is a pretty easy one to keep track of.
You can also choose to honor-system it and if you know in your heart that you saw that one dragonfly, that’s good enough.
Wild vs. domestic
I suggest only playing with wild observations. It doesn’t have to be a “native” species – it can be a weed, feral, etc – and I understand that there are edge cases, but try to use “a person did not place this here on purpose and it’s not clearly an escapee from the garden six inches away” as a heuristic.
(But if you’re playing in a very urban area and want to study, idk, trees, you might not have that many, say, wild trees available. Most urban parks are planted on purpose. You can choose something else for criteria – just maybe think about it in advance.)
I really recommend not counting zoos, botanical gardens, pet shops, or other places designed to put a lot of rare species all in the same space. Your SPECIESQUEST is your own, however.
Decide how long your game will last for. You can do a shorter one – or maybe arrange shorter “sprints” within your longer game. I am planning to play over the course of a year.
PLAY
Go out and document some guys.
Note:
People CAN join partway through the session, or dramatically switch their goals. They’ll be at a disadvantage, of course.
SCORING:
Local %
At the end of the time period, everyone determines how many SPECIES on their CHECKLIST they observed. Report your score as a %.
Total Species
Bigger number = more victory.
Crowning Victors
In theory, all the Local % players should be able to compete directly against each other – highest % wins. All the Total Species players should be able to go head to head with others playing in their categories (“Most Bird Species Seen”, etc.)
In practice, probably some of the categories are way harder than others – the choose-your-own-approach is meant to deal with this by letting you set your own limits, but maybe you have a player who is like really into mammals and deems this setback an acceptable price for motivation to go look for mammals, and only identified 4/10 species of weasels that live in their region, but you want to acknowledge them anyhow because that’s still a pretty impressive number of weasels to see, let alone identify. Maybe none of your Total Species players have the same categories. Maybe one of your crew was technically a Local % player but made an impressive showing at total iNaturalist observations over the year… I suggest handing out trophies liberally.
(If you DON’T want to be generous handing out trophies, tailor your SPECIESQUEST league so that everyone is playing with the same ruleset, or something.)
Note:
You can just play on your own, without a league, as a personal challenge.
If you find a species that is unknown to science, that counts for 10 observations for scoring. But you have to be really sure that it’s actually new.
The real prize is the guys we found along the way.
Go out and enjoy SPECIESQUEST 2026. Let me know if you’re playing and/or starting a league with your own friends.
If you live in this one tiny county in California, you might be more likely to die from Sin Nombre Virus than in a car crash.
In the same way that “why does the frozen spinach I want to buy cost much more than it used to?” engages with a vast interconnected web of economies and monetary policies and farmers and supply chains, asking “what’s up with this rare disease people sometimes get in my part of the world?” is actually a question about the entire ecosystem, plus how organisms even work.
The reason you have to think about the natural world when you do biosecurity is that the vast majority of human diseases come from animals. What we think of as diseases to humans is a two-dimensional slice of a giant, rotating, obscure shape of many dimensions – a whole world of diseases, little communities of microbes and macrobes interacting and evolving and getting sick and occasionally passing their diseases around between them. Communities of parasites built on communities of hosts, all colliding constantly. This is the large scale of biosecurity. Nothing in infectious disease research makes sense without it. Any question about human health or symptomology or individual risk or what have you is a tiny speck on the shore of this ocean.
Occasionally, one of those parasites reaches out of the host community it’s adapted to, and finds a foothold in another host. And so the sphere gets a little bigger, a little more interconnected.
Today we’ll be looking at a single slice of the grand pageant, about this size – one virus in one part of the world, that sometimes slips from its home and finds its way into a human animal.
Sin Nombre Virus
Sin Nombre Virus was first characterized in 1993 in New Mexico. Since then, there haven’t been many identified infections, but every now and then, cases crop up. Even in the medically well-equipped United States, Sin Nombre virus has maintained an astonishing 40% mortality rate.
Here’s a map of hantavirus infections in the US by state, since its discovery. We can see that it’s far-reaching, but it clearly has a geographic localization.
So if you live in California, your risk is even comparatively low. But out of curiosity, let’s look closer at a map of SNV infections in California counties.
Huh, what’s the deal with that one county? Note that this is a total case map, not a per capita case map, and that county doesn’t have any large cities. In fact, it’s the 4th least populated out of California’s 58 counties. So the risk is even higher than that map makes it look!
But it’s pretty unlikely that any given blogger would live in that county, isn’t it?
Ha ha, what a funny idea. Anyway, I happened to take an interest in this rare, hyperdeadly disease.
The virus without a name
Most current reporting describes the disease we’re looking at today with the more general name of hantavirus – which it is, but there are multiple human diseases in the hantavirus family.
They’re split into the Old World and New World hantavirus. The Old World hantaviruses cause hantavirus hemorrhagic fever with renal syndrome (HFRS) in Eurasia.
The New World hantaviruses include our subject of interest today as well as the related Andes virus in South America (plus a few other, even rarer North American viruses we’ll discuss later). Andes virus has similar symptoms and is about as deadly as Sin Nombre Virus, but it sees more cases every year – 100-200 versus North America’s “dozens” – and it shows occasional person-to-person spread. We’ll come back to that, but for now, I’m focusing on the most common North American hantavirus because it’s the one that’s in my own backyard. …Potentially literally.
The North American hantavirus we’re discussing today is more specifically known as Sin Nombre virus. Why is it called that?
It was discovered in 1994 after a lot of people got sick in the Four Corners region of New Mexico. Local Native communities actually had stories about odd numbers of people getting suddenly sick and dying during years where the pine nut harvest was good, and indeed, 1994 was a good pine nut mast year. Because of abundant nuts to eat, the mouse population exploded and came into a lot of contact with humans, and enough people got sick and died that USAMRIID and the CDC investigated. And they found a virus at the root.
Ongoing practice at the time was to name newly-discovered viruses after geographic locations nearby the site of origin. But this was already facing pushback – who wants to take a vacation to the scenic Ebola River? On top of that, the area and early cases were heavily Native American communities, and before the disease was shown to NOT be communicable, Native groups were facing racism and shunning over this mystery disease.
The Four Corners region didn’t want it to be the Four Corners virus; the nearby Muetro Canyon was proposed but rejected because the Navajo community didn’t want more stigma (and also Muerto Canyon was named after a massacre against the Navajo), and back and forth, and eventually they just called it the virus without a name, AKASin Nombre virus.
I have some thoughts on infectious disease naming that are too long for the current margin to contain, but I will say that I think this is the kind of cool infectious disease naming schema that you can pull off once.
Mice
This is the western deer mouse, Peromyscus sonoriensis. Sin Nombre virus lives here.
The worst part of biosecurity is having to look at something like this and be like “this thing is the enemy.” Okay, maybe that’s not the worst part.
This is a pretty common strategy of infectious viruses – playing the slow, long game. Humans have a few: cytomegalovirus, herpes simplex virus (especially HSV-1), Human T-cell lymphotropic virus type 1… viruses that lots of people have for their entire lives, and have no idea that they have.
Compare also things like the common cold or human papillomaviruses that cause warts – shorter lifespan and some chance of symptoms but also not much, really. The immune system eventually clears these out in most cases without help, but they have time and means to spread, and they circulate among us and periodically annoy us, but mostly, they don’t kill us.
The deer mouse is not the same thing as the house mouse Mus musculus, which you’re probably more familiar with. But let’s take a minute here.
There’s mice and then there’s mice
We all know Mus musculus – it’s the common house mouse, which has spread worldwide alongside people. If humans build a town, the house mouse will soon follow. There are a lot of less-common related species of mice, like the adorable African pygmy mouse (Mus minutoides).
But Peromyscus sonoriensis isn’t either of these. Technically speaking, is it a rat or a mouse?
Well, what a great question. It’s neither.
Huh, you might think. Mice are on there twice? If you know your way around a phylogenetic tree, you may wonder: maybe the common ancestor was more like a mouse, and it’s rats that are doing something weird?
Ha. Haha. Hahahaha. No. The real situation is more complicated than you could possibly believe.
Rats and mice have evolved multiple times, with some incredibly weird variations in the mean time.
This is the distance between the western deer mouse and the house mouse:
Despite all of this genetic distance, hice mice and deer mice occupy extremely similar niches. Where the western deer mouse is native, it’s completely comfortable cozying up to human dwellings and making its nests inside our big, fancy, warm, dry, food-filled nests.
And deer mice that are widely regarded as the vectors of Sin Nombre virus – the host species that it’s evolved to circulate in. In New Mexico, two studies (one statewide, one in an area where a human was infected) found that about 35% of deer mice had the virus at any given time. Eyeballing it, this lines up pretty well with a “disease circulating stably among the mouse population that rarely spontaneously spills into humans” situation.
…But wait, are deer mice really the only carriers? That second study also found replicating, viable Sin Nombre virus in other local rodents – including the house mouse, mus musculus! The sample sizes weren’t huge, but 3 out of the 9 captured had it!
Note, however, that they only found house mice at one of the sites. There were many more deer mice than house mice. But still, 3/9!
What I don’t know, and what I don’t think anyone knows, is the degree to which hantavirus actively circulates among these other rodents. Are they just getting it incidentally from neighboring deer mice, or do they pass the virus around between themselves too? Is Sin Nombre virus just as at home in them as it is in western deer mice?
The literature is very clear that deer mice are the ones associated with Sin Nombre virus infection. For instance:
The most common hantavirus that causes HPS [that virus being SNV] in the U.S. is spread by the deer mouse.
But “common house mice (Mus musculus), which are prevalent in urban and suburban communities, do not carry hantavirus,” said Charles Chiu, MD, PhD, professor of laboratory medicine in the division of infectious diseases at the University of California, San Francisco.
(Sidenote: this article also quotes one of those New Mexico survey articles I mentioned above, saying that it “found less than 9% of deer mice had the virus.” The study did report that 10/113 deer mice had antibodies to SNV, but it also found that 37/113 of the deer mice had SNV DNA in their system. This is weird, because viral DNA is a sign of an active infection – it’s made by a virus! – but the immune response can linger for a long time after infection, so we’d really expect more mice to have antibodies to SNV than to have SNV DNA. The study does mention that this trend held up across all rodents studied, so maybe this just has to do with the sensitivity of their antibody assay.)
And there are a lot of cases where people got sick, in which the victims knew they’d come into contact with material contaminated by deer mice.
But there are also cases of infection where nobody saw a mouse, and the presence of any mice at all just has to be intuited. Is it possible that Mus musculus is responsible for some Sin Nombre cases in humans?
The public health literature is pretty unanimous about the deer mouse thing, so I’m going to proceed assuming that’s effectively the only way any human gets Sin Nombre virus, but I don’t understand why they’ve ruled out other mice too.
Human
The adventures of a dead-end host
Sin Nombre virus is a transient inside human beings – it’s not adapted here, it doesn’t stay here. We know this because when we isolate the virus from infected humans, it doesn’t easily reinfect deer mice. This suggests that small mutations have to occur to make the virus able to replicate in humans – ones that make it less viable within mice.
[…] which implies that humans are truly dead-end hosts of SNV. Thus, virus evolution is primarily, if not exclusively, occurring in the natural rodent reservoirs.
But SNV can infect humans, and a virus has to replicate to make its host sick. How does it do that?
Well, it’s almost always inhaled from mouse-contaminated material. Then the virus somehow gets into the blood stream.
Once it’s there, Sin Nombre virus replicates inside a variety of human cells, but especially likes endothelial cells and macrophages.
Endothelial cells are the guys that line our blood vessels. They grow everywhere the blood vessels grow, which is to say, all over
Macrophages are a kind of immune cell that devours pathogens. The SNVs are captured by the macrophages, and as with all of their prey, are moved into a lysosome – a cellular chamber that turns into an acid bath, designed to inactivate complex biomolecules (and pathogens they’re attached to) trapped within. But the SNV particles escape into the cell membrane just as the acidification starts.
Replicating inside immune cells is a pretty common strategy for viruses. Sure, the immune cells try to spot and destroy pathogens, but they also end up capturing and moving pathogens around a lot, which can be a big boon if the pathogen has a way to just not get killed by the cell.
Some macrophages roam the bloodstream, but others are concentrated in outposts around the body. Some are in the lungs.
As far as I can tell, Sin Nombre Virus probably gets into the lungs, then infects the alveolar macrophages (and possibly other lung-based immune cells), and then escapes from those into the blood stream where it might infect other endothelial cells. They might also manage to get through tears or thin spots in the alveolar-capillary membrane and get straight into the blood – that’s just a guess.
Replicating in endothelial cells seems kind of overpowered for a virus, right? Like, we have a gazillion of ‘em and they’re all over the body and once you’re next to the bloodstream, it’s an easy highway for a virus to get from one part of the body to a totally different part of the body. to spread from one part of the body to a totally different part of the body – and if you mounted an inflammation or severe immune response, that seems like that would kill the entire host easily and quickly.
And indeed, Sin Nombre Virus does kill its host quite effectively. Ebola, another famously lethal disease, also replicates in endothelial cells. Covid seems to be able to sometimes (in addition to its main habitat in the respiratory tract, an interesting similarity between it and Sin Nombre Virus.)
So is replication in endothelial cells a sure sign that a disease will wreck havoc on the human body?
Well, no. Dengue fever replicates in endothelial cells, and most of its hosts are asymptomatic or mildly symptomatic. Its fatality rate is literally one in a million. And moreover, cytomegalovirus is an endothelial replicator. Like we talked about before, cytomegalovirus of those viruses that’s almost a commensal – most people have symptomless cytomegalovirus infections. (It can cause disease in unborn fetuses, infants, and the immunocompromised, and seems to contribute to cancer risks down the line – it’s not great – but, again, most people have it.)
Also, lots of viruses attack tissues that are essential and would be bad to call the full attention of the immune system to – herpes viruses (another near-commensal genre of virus that most infected carry without any symptoms whatsoever) infect nerve cells, for instance. Lots of viruses infect the lungs, which are famously important, and some of them kill you and some of them are no big deal.
So I think a general lesson here is that the driver of virulence here has more to do with the rate of growth / level of viruses active at once and the degree to which they activate the immune system, not the infected tissue.
Do a bunch of people within the regions where it is have indications of asymptomatic or past infections?
This is a great question. After all, mice have it quietly, and people seem to have the capacity to carry or fight off a lot of infections quietly without notable symptoms. Are we sure this isn’t the case for hantavirus?
Well, so far as I know, nobody has checked.
Wait, can we talk about the actual disease?
Yeah, fine I guess.
According to the CDC, the early symptoms of Sin Nombre virus disease in humans – AKA hantavirus pulmonary syndrome (HPS) or hantavirus cardiopulmonary syndrome (HCPS) – emerge 1-8 weeks after acquiring the virus. They start out, like a lot of fucked up viral diseases, with generic symptoms:
Muscle aches
Fever and chills
Malaise
Headaches
Abdominal pain
Though “aches” might be a standout. University of Colorado Health (Colorado has a lot of SNV cases) reports that severe muscle aches, especially in the back and lower extremities, are a common hallmark of HCPS cases. (Hey, I got severe leg pain when I got shigellosis on purpose too – shigellosis, much like Sin Nombre virus, is an infectious disease that notably does not target the legs. What’s up with that?)
4-10 days after this, the cardiopulmonary stage of disease begins, AKA “the part that kills you”:
Coughing
Shortness of breath
Fluid buildup in lungs/chest
Tachycardia
Arrythmia
Cardogenic shock
Respiratory failure
HCPS has a 40% death rate. Deaths occur 24-48 hours after the start of the cardiopulmonary phase. There is no vaccine or known effective antiviral.
Buying time
If you get HCPS and reach the cardiopulmonary stage, the thing that will save your life is a medical technology called extracorporeal membrane oxygenation (ECMO). An ECMO device draws large volumes of blood out of the body via inserted tubes (called cannulae), runs the blood through an artificial lung (called a membrane oxygenator) to remove carbon dioxide and reoxygenate the blood cells, and puts the blood back in the body.
HCPS seems to be one of those diseases where the body can rally and fight off the disease, if it has enough time. I attended an online lecture delivered by clinician Dr. Greg Mertz and this is the sense I got: SNV doesn’t permanently damage the heart and lungs, it just overwhelms them. If ECMO takes over while the heart and lungs are out of commission and keeps plenty of oxygenated blood in the system, the immune system can finish the job and the heart and lungs can go back to work afterward.
If you go to a hospital with symptoms and they make a presumptive diagnosis of HCPS, you can opt into having the ECMO cannulae inserted in advance – they won’t start ECMO until you go into shock (because your heart/lungs fail), but if you do go into shock, they’ll be able to start re-oxygenating your blood immediately. At this point, doing this changes your odds of survival from 50% to 80%.
(I see that in my notes from that talk, I also wrote “Do not go into shock”, as it leads to “DEATH V FAST.” So if you get to decide at some point whether or not to go into cardiac shock in general, try not to.)
So if you think you’ve been exposed to SNV and 1-9 weeks later you start experiencing arrhythmia and shortness of breath, proceed straight to a hospital with an ECMO device.
ECMO devices are not extremely common. You can find out which hospitals near you have ECMO devices on the Extracorporeal Life Support Organization website. If you happen to be reading in Mono County, your nearest ECMO is probably in Reno Renown Regional Medical Center.
…But if you’re one of the 12,000 residents of Mono County, then yeah, probably. Mono County has had an unusually high 3 HCPS deaths from hantavirus this year, so you have a 0.025% chance of dying from HCPS.
You might actually be more likely to die from hantavirus as in a car crash (0.012% chance in any given year.)
(Sidenote: Naively, I’d expect Mono County residents to have a 0.000004% of dying by being struck by lightning like anyone else, but if you actually look into it, Florida specifically and the southeast generally have a really disproportionate number of lightning strike deaths. We should probably stop rhetorically treating getting struck by lightning as an entirely random act of god and start thinking of it as a physical event with contributing factors like everything else.)
Questions
Why is the geographic range of hantavirus infection so limited?
Let’s go back to that map of USA state-level infections.
So mostly, that makes sense. But how are there ANY cases in the east half of the state?
SNV’s weird siblings
Those other HCPS cases on the east coast? Well, they’re not (or at least, not only) people who happened to travel from the West Coast, and they’re not (or at least, not only) far-ranging western deer mice.
Those are the work of other, rarer hantaviruses, carried by other rodents, spilling over occasionally into other humans in the same way, causing HCPS, and with about the same fatality rate.
Each of these is really rare, even rarer than SNV. But that’s odd in and of itself, right? Like, do all of these host species just interact less often with humans than deer mice in the Western US? Are the viruses less common in their hosts, or even less transmissible than SNV? The answers might be out there, but I don’t know that they are.
I ’m also curious about the California county-level breakdown: why Mono County? (And note that this is raw cases, not cases per capita – Mono County has a tiny population.) Is it because there are more deer mice? Or is hantavirus localized to certain populations of deer mice?
Well, here’s this other data on seroprevalance of hantavirus among captured mice in various counties. Sure enough, Mono County has the highest seroprevalance, at 31%, but apparently 25% of tested mice in Santa Barbara County also had SNV, and Santa Barbara has a lot of people in it!
So why does Santa Barbara see very few human cases, while Mono County has a lot?
Here’s my guess at why: it has to do with the houses, and it has to do with mice. Mono County has a lot of barns, sheds, and vacation houses that are left empty part of the year. The classic situation where a person gets SNV is cleaning out a shed or outbuilding that’s been inhabited by mice, kicking up a lot of mousy dust and particles, and inhaling SNV. A shed or a building that’s left for the summer or winter is a nicer place to build a shelter than under a bush, but it’s still not that cozy – it might not have food inside so the mouse still has to forage a lot, and it might get very cold or very dry. There might not be many other buildings nearby. A region-adapted, mostly-wild deer mouse, is going to have a better go in an outbuilding then the urban Mus musculus – and indeed, every mouse I’ve seen or caught around my home has been a deer mouse.
Santa Barbara County is much more urban and has a warmer climate. I bet the mice that people encounter there are almost all Mus musculus. I bet all the Santa Barbara deer mice live in the wild, outcompeted in cities by the larger and more urbanized mus musculus.
And the deer mice are god’s chosen carriers of SNV, and the Mus musculus aren’t. It’s just a deer mouse disease. So it’s much more likely to crop up where people interact with deer mice, and they do so a lot more in these rural, more-wild environments.
It’s an apparent puzzle that makes a lot of sense once you just ignore the human health angle for a second. SNV is a deer mouse disease that circulates among deer mice. Think about which mice want to live where. Humans, as is often the case, are providers and users of nests, and otherwise, are only relevant incidentally.
But wait, can we check this?
If my model is correct, areas that have high SNV caseloads will:
Be mostly rural (probably without major cities?)
Have extreme climates
Have a lot of outbuildings, plus homes that are inhabited seasonally
It would also be interesting if they’re clearly geographically clustered – like if specifically one part of the world is a hantavirus hotbed.
Yeah, let’s look at some other states that get a lot of SNV cases. I don’t expect to get great data at anything lower than the county level. Colorado and New Mexico both get more SNV cases than California, and have county level data.
I tried to look into this further, and ran into kind of a dead end. Or maybe I’m just wrong.
The counties with the highest rates include La Plata and Weld counties in Colorado, and Mckinley county in New Mexico, which is such a standout that it dwarfs the others.
La Plata County has a population of 55,638 with the largest city (Durango) at 10,000. It has some parks and overlaps a national forest, no major ski areas.
Weld County has several cities and a population of 329,000. (It contains parts of some large cities that are on the border so it’s hard to break down for sure, but a lot of people live here.) Okay, not looking great. It’s fairly flat with some mountains, and mostly farming country.
Mckinley county has one city of 20,000 and no other cities, but a lot of smaller towns and census-designated places and such. Its total population is 73,000, which is pretty big! I can’t find indications that it has a lot in the way of seasonal dwellings – there aren’t many ski resorts. The county does seem to be pretty dispersed, housingwise, which might imply more outbuildings.
So, uh, none of this actually cleanly supports my mode, but it’s not necessarily evidence against it either. We might just need data on which kinds of mice are common in human dwellings in these areas, and how common mice are overall.
What makes Andes virus infectious interpersonally, and SNV not?
It seems like ANDV builds up in the salivary glands of humans, and saliva is its mode of transmission. SNV doesn’t do that.
SNV collects in the lungs and heart. ANDV collects in the heart, lungs, and salivary glands, and tests studies have indicated virus in fluids from both of these. It seems to spread via saliva, droplets, and aerosols. Sex and close contact are major risk factors. Otherwise, ANDV has a similar features and fatality rate to SNV.
Saliva also seems to be how deer mice spread SNV and ANDV among each other – both of them show up in the salivary glands of their host mice, but only ANDV inhabits human salivary glands.
(If we could somehow prove that ANDV could spread from the lungs, well, that would suggest some new mechanism also in play – but that seems hard to test, given that the mouth is, you know, between the lungs and the rest of the world.)
That said, I actually don’t understand why ANDV isn’t airborne, or otherwise transmitted from the lungs. The mousy particles that infect humans seem to be from kicked up dust and such, so there’s reason to think it could be aerosolized – maybe the virus particles don’t escape the lungs very well?
That’s all the things I know about Sin Nombre virus, plus some things I don’t. Let me know if you have the answers. In the mean time, don’t die of cardiopulmonary shock. I, for one, am doing my best out here.
Remember early 2020 and reading news articles and respected sources (the WHO, the CDC, the US surgeon general…) confidently asserting that covid wasn’t airborne and that wearing masks wouldn’t stop you from catching it?
Man, it’s embarrassing to be part of a field of study (biosecurity, in this case) that had such a public moment of unambiguously whiffing it.
I mean, like, on behalf of the field. I’m not actually personally representative of all of biosecurity.
I did finally grudgingly reread my own contribution to the discourse, my March 2020 “hey guys, take Covid seriously” post, because I vaguely remembered that I’d tried to equivocate around face masks and that was really embarrassing – why the hell would masks not help? But upon rereading, mostly I had written about masks being good.
The worst thing I wrote was that I was “confused” about the reported takes on masking – yeah, who wasn’t! People were saying some confusing things about masking.
I mean, to be clear, a lot of what went wrong during covid wasn’t immediately because biosecurity people were wrong: biosecurity experts had been advocating for years for a lot of things that would have helped the covid response (recognition that bad diseases were coming, need for faster approval tracks for pandemic-response countermeasures, need for more surveillance…) And within a couple months, the WHO and the Surgeon General and every other legitimate organization was like “oh wait we were wrong, masks are actually awesome,” which is great.
Also, a lot went right – a social distancing campaign, developing and mass-distributing a vaccine faster than any previous vaccine in history – but we really, truly dropped the ball on realizing that COVID was airborne.
In his new book Air-borne: The hidden history of the air we breathe, science journalist Carl Zimmer does not beat around this point. He discusses the failure of the scientific community and how we got there in careful heartbreaking detail. There’s also a lot I didn’t know about the history of this idea, of diseases transmitting on long distances via the air, and I will share some of it with you now.
Throughout human history, there has been, of course, a great deal about confusion and debate about where infectious diseases came from and how they were spread, both before and to some extent after Louis Pasteur and Robert Koch et al illuminated the nature of germ theory. Germ theory and miasma theory were both beloved titans. Even after Pasteur and Koch had published experiments, the old order, as you may imagine, did not go quietly; there were in fact series of public debates and challenges with prizes and winners that pitted e.g. Pasteur up against old standouts of miasma theory.
One of the reasons that airborne transmission faced the pushback it did is that it was seen as a waffley compromise of a return to miasma theory. What, like both a germ and the air could work together to transmit a disease? Yeah, sure.
Airborne transmission was studied extensively in the 1950s. It eventually became common knowledge that tuberculosis was airborne. That other diseases, like colds and flu and measles, could be airborne, was the subject of intense research by William and Mildred Wells, whose vast body of work included not only proving airborne transmission but experimenting with germ-killing UV lights in schools and hospitals — and who remain virtually unknown to this day.
Let us acknowledge a distinction often made between droplet-borne diseases, where heavy wet particles might fly from a sneeze or cough for some six feet or so, to airborne diseases, which might travel across a room, across a building, wafting about in the air for hours, et cetera. This distinction is regularly stressed in the medical field although it seems to be an artificial dichotomy – spewed particles seem to be on a spectrum of size and the smaller ones fly farther, eventually becoming so small they’re much more susceptible to vagaries in air currents than to gravity’s downward pull. Droplet-borne diseases have been accepted for a long time, but airborne diseases were thought by the modern medical establishment to be very rare.
(I forget if Zimmer makes this point, but it’s also easy to imagine how it’d be easier for researchers to notice shorter-distance droplet-borne transmission – the odds a person comes down with a disease relates directly to how many disease particles they’re exposed to, and if you’re standing two feet away from a coughing person, you’ll be exposed to more of the droplets from that blast than if you’re ten feet away. Does that make sense? Here’s a diagram.)
Aerosols disperse from their source over distances.
(But that doesn’t mean that ten-foot transmission will never happen. Just that it’s less likely.)
Why didn’t the Wells’ work catch on? Well, it was controversial (see the ‘return to miasma’ point above’), and also, they were just unpleasant and difficult to work with. They were offputting and argumentative. Also Mildred Wells was clearly the research powerhouse and people didn’t want to hire just her, for some reason.* Their colleagues largely didn’t want to hire and fund them or to publish their work. We have a cultural concept of lone genius researchers, but these are, in terms of their impact, often fictional – science is a broadly collaborative affair.
The contrast in e.g. Koch and Pasteur’s status vs. William and Mildred Wells made me think about the nature of scientific fame. I wonder if most generally-famous scientists were famous in their lifetimes too. Koch and Pasteur were. Maybe most famous scientists are also famous because they’re also good science communicators. I’m sure that also interplays with getting your ideas out into the world – if you can write a great journal article that sounds like what you did is a big deal, more people will read it and treat it like a big deal.
The Wells were not a big deal, not in their day nor after. Their work, studying disease and droplet transmission and the possibility of UV lamps for reducing disease transmission (include putting lamps up in hospitals and schools), struggled to find publication and has only recently been unearthed as a matter of serious study.
Far UV lamps are the hot new thing in pandemic and disease response these days. Everyoneistalkingaboutthem.
There’s variations and nuance, but the usual idea works like this: you put lamps that emit germ-killing UVC light up in indoor spaces where people spend a lot of time. UVC light can causes skin cancer (albeit less than its higher-energy cousin, UVB). But you can just put the lamps in ventilation systems or aimed up at the ceilings, where they don’t point at people or skin but instead kill microbes in the air that wafts by them. Combined with ventilation, you can sterilize a lot of air this way.
William and Mildred Wells found results somewhere in between “positive” and “equivocal” – the affect being stronger when people spent more of their day under the lamps, e.g., pretty good in hospital wards and weaker in schools.
They’re not too expensive and could be pretty helpful, especially if they became de facto in places where people spend a lot of time – and especially in hospitals. Interest in this is increasing but there’s not much in the way of requirements or incentives for any such thing yet.
*Sexism. Obviously the reason is sexism.
The other heroes of the book are the Skagit Valley Chorale. In March 2020 a single Skagit Valley Chorale choir rehearsal transmitted multiple fatal covid cases during a single choir practice. Afterwards, the survivors worked with researchers, who figured out where everyone was standing, where points of contact were, did interviews and mapping and figured that there had been no coughing or sneezing, that the disease had in fact been flung at great distances just by singing – that it was really airborne. (There were other studies in other places indicating the same thing.) But this specific work of contact tracing was a focus and was instrumental and influential, and cooperation between academic researchers and these grieving choir members formed an early, distinct piece of evidence that covid was indeed airborne.
I think being part of research like this – an experimental group, opting into a study – is noble. It’s selfless, and what a heroic and beautiful thing to do with your grief and your suffering, to say: “Learn everything you can from this. Let what happened here be a piece in the answer to it not happening again.”
(Yeah, I got dysentery for research, but listen, nobody in the Skagit Valley Chorale got $4000 for their contributions. They just did it for love. That’s noble.)
There was also a cool thread of the story that involved microbiologists like Fred Meier and their interactions with the early age of aviation – working with Lindbergh and Earhart and balloons and the earliest days of commercial aviation to strap instruments to their crafts and try to capture microbes whizzing by.
And they found them – bacteria, pollen, spores, diseases, algaes, visitors and travellers and tiny creatures that may have lived up their all their lives. Another vast arm of the invisible world of microbes.
I’ve been interested in the mechanics of disease transmission for almost as long as I’ve been interested in disease. In freshman year in college I tried an ambitious if bungled study on cold and flu transmission in campus dorms. (That could have been really cool if I’d known more about epidemiological methods or at least been more creative about interpreting the data, I think. Institutions are famously one of the easier places to study infectious diseases. Alas.) Years later I tried estimating cold and flu transmission in more of an EA QALY/quantifying-lost-work days sense and really slammed into the paucity of transmission studies. And then covid came, and covid is covid – we probably got the best data anyone has ever gotten on transmission of an airborne/dropletborne disease.
More recently, I’ve been doing some interesting research into rates and odds of STD transmission, and there’s a lot more there: there’s a lot of interest and money in STD prevention, and moreover, stigmatized as they are, it’s comparatively easy to determine when certain diseases were caught. They transmit during specific memorable occasions, let’s put it like that.
For common air- or droplet-borne diseases? Actual data is thin on the ground.
I think this is one of the hard things about science, and about reasoning in and out of invisible, abstract worlds – math, statistics, physics at the level of atoms, biology at the level of cells, ecology at the level of populations, et cetera. You know some things about the world without science, like, you don’t need to read a peer-reviewed paper to know that you don’t want to touch puke, and you don’t need to consult with experts in order to cook pasta. The state of ambient knowledge around you takes care of such things.
And then there’s science, and science can tell you a lot of things: like, a virus is made of tiny tiny bricks made of mucus, and your body contains different tiny virus detectors (also themselves made of mucus), and we can find out exactly which mucus-bricks of the virus trigger the mucus-detectors in your body, and then we can like play legos with those bricks and take them off and attach them to other stuff. We know about dinosaurs and planets orbiting other stars.
And science obviously knows and tells us some useful stuff that interacts with our tangible everyday world of things: like, you can graft a pear tree onto a quince tree because they’re related. A barometer lets you predict when it’s going to rain. You can’t let raw meat sit around at room temperature or you might get a disease that makes you very sick. Antibiotics cure infections and radios, like, work.
And then there’s some stuff that’s so clearly at this intersection that you might assume it’s in this domain of science. Like, we know how extremely common diseases transmit, right? Right?
It used to blow my mind that we know enough about blood types to do blood transfusions and yet can’t predict the weather accurately. Now it makes visceral sense to me, because human blood mostly falls into four types relevant to transfusions, and there are about ten million factors that influence the weather. (Including bacteria.)
Disease transmission is a little bit like predicting the weather, because human bodies and environments are huge complicated machines, but also not as complicated, because the answer is knowable – like, you could do tests with a bunch of human subjects and come up with some reasonable odds. We just… haven’t.
Actually, let’s unpack this slightly, because I think it’s easy to assume that airborne (or dropletborne) disease transmission would be dirt cheap and very easy to study experimentally.
To study disease transmission experimentally, you need to consider three things (beyond just finding people willing to get sick):
First, a source of infection. If you’re trying to study a natural route of infection like someone coughing near you, you can’t just stick people with a needle that has the disease – you need a sick person to be coughing. For multiple reasons, studies rarely infect a person on purpose with a disease, let alone two groups of people via different routes (the infection source and the people becoming infected) – you might need to find a volunteer naturally sick with the disease to be Patient Zero.
Second, exposure. People are exposed to all sorts of air all the time. If you go about your everyday life and catch a cold, it’s really hard to know where you got the cold from. You might have a good guess, like if your partner has a cold you can make a solid statistical argument about where you were exposed to the most cold germs – or you might have a suspicion, like someone behind you on the bus coughing – but mostly, you don’t know. A person in a city might be exposed to the germs of hundreds on a daily basis. In a laboratory, you can control for this by keeping people isolated in rooms with individually-filtered air supplies and limited contact with other people.
Third, when a person is exposed to an infectious disease, it takes time to learn if they caught it or not. The organism might get fought off quickly by the body’s defenses. Or the organism might find a safe patch of tissue to nestle in and grow and replicate – the incubation period of the infection. It’ll take time before they show symptoms. Using techniques like detecting the pathogen itself, or detecting an immune response to the pathogen, might shave off time, but not a lot, you still have to wait for the pathogen to build up to a detectable level or for the immune response to kick in. Depending on the disease, they also may have caught a silent asymptomatic infection, which researchers only stand a chance of noticing if they’re testing for the presence of the pathogen (which depending on the pathogen and the tests available for it, might entail an oral or nasal swab, a blood test, feces test…)
So combine these things – you want to test a simple question, like “if Person A who is sick with Disease X coughs ten feet away from Person B, how likely is Person B to get sick?” The absolute best way to get clean and ethically pure data on this is to find a consenting Person A who is sick with the flu, find a consenting Person B (ideally who you are certain is not already sick, perhaps by keeping them in an isolated room with filtered air beforehand for the length of the incubation period), have Person A stand ten feet away and cough, and then sweep Person B into an isolated room with filtered air for the entire plausible incubation period, and then see if they get sick, and then have this sick person cared for until they are no longer infectious.
And then repeat that with as many Persons B as it takes to get good data – and it might be that only, like, 1% of Persons B get sick from a single sick person coughing 10 feet away from them. So then you need, I don’t know, 1000 Persons B at least to get any decent data.
It’s not impossible. It’s completely doable. I merely lay this out so that you can see that producing these kinds of basic numbers about disease transmission would instantly entail a lot more expense and human volunteers than you might think.
A friend of mine did human challenge trials studying flu transmission, and they did it similarly to this – removing the initial waiting period (which is fair, most people are not incubating the flu at any given moment) and with more intense exposure events, with multiple Persons B in a room actively chatting and passing objects around with a single Person A for an hour, and then sending Persons B to a series of hotel rooms for a few days to see if anyone got sick.
(What about going a step further: just having Person A and Persons B in a room, Person A coughs, and then send Persons B home and call them a few days later to ask about symptoms? You could compare this to a baseline of Persons C who were not in a room with a Person A coughing (“C” for “control”). Well, I think this would get you valid and usable numbers, but exposing people to infectious diseases that could then be freely passed on to nonconsenting strangers is considered a “bioethics no-no” – and so researchers have, to my knowledge, mostly not tried this.)
(Maybe someone did that in the sixties. That seems like something they’d have done back then.)
The point is, it’s like, expensive and medium hard to study airborne disease transmission experimentally. Adjust your judgment accordingly.
Anyway, fascinating book about the history of the history of that which you think might be better understood by virtue of being a life-and-death matter millennia old, but which is, alas, not.
Here are some questions I was left with at the end of the book:
What influences whether pathogens are airborne-transmissible? Does any virus or spore coughed up from the lungs have about the same chance of becoming airborne, or do other properties of the microbe play a role? (I was hoping the book would explain this to me, but I think the research here may not exist.)
Zimmer is clearly pro-far-UV but the Wells’ findings on far UV lamps in schools was in fact pretty equivocal – do we have reason to think current far UV would fare better? (I know I linked a bunch of write-ups but I’m not actually caught up on the state of the research.)
Some microbes travel for long distances, hundreds of miles or months, while airborne. Often high in the earth’s atmosphere. How are these microbes not all obliterated by solar UV?
This summer, I participated in a human challenge trial at the University of Maryland. I spent the days just prior to my 30th birthday sick with shigellosis.
What? Why?
Dysentery is an acute disease in which pathogens attack the intestine. It is most often caused by the bacteria Shigella. It spreads via the fecal-oral route. It requires an astonishingly low number of pathogens to make a person sick – so it spreads quickly, especially in bad hygienic conditions or anywhere water can get tainted with feces.
It kills about 70,000 people a year, 30,000 of whom are children under the age of 5. Almost all of these cases and deaths are among very poor people.
The primary mechanism by which dysentery kills people is dehydration. The person loses fluids to diarrhea and for whatever reason (lack of knowledge, energy, water, etc) cannot regain them sufficiently. Shigella bacteria are increasingly resistant to antibiotics. A disease easily treatable by lots of fluids and antibiotics is becoming more lethal.
Can someone do something?
The deal with human challenge trials
Clinical trials in general are expensive to run but pretty common; clinical trials where you are given the disease – “challenged”, AKA “human challenge trials” – are very rare. The regular way to investigate a possible treatment is to make a study plan, then find people who have the disease and offer to enroll them in the experimental treatment. Challenge trials are less common, but often more valuable for research – shigellosis is a fast-acting disease that is imminently treatable by antibiotics and uncommon in the US. It would be very difficult to test an alternative shigellosis treatment in the US in the conventional way, but it’s a great candidate for challenge trials.
I’d signed up for email alerts on upcoming challenge trials at the nearby University of Maryland, and got one about an upcoming study. It caught my eye that it was for a phage-based treatment. Bacteriophages are really promising antibacterial medicines, not to mention what I’d studied as an undergrad.
Here’s the thing: you really only get good medical research out of human subjects. Also, I could use $4000 and this seemed like a cool way to spend a couple weeks and help out medical research. So I signed up, got a check-in general health appointment, and shortly after, was told I was in. I made plans to spend my 30th birthday in a dysentery ward.
Dysentery: it’s a modern disease
Many of you reading this will know about dysentery from the 1971 simulation game The Oregon Trail (or its later versions). The actual Oregon Trail was a network of trails and the corresponding migration of mostly-white pioneers, moving on foot and on ox-drawn wagon from the eastern US to the western US between 1830 and 1869. About 400,000 people* crossed the Oregon Trail in this period, and a lot of them were on similar trails – a bunch of stressed and malnourished people, traveling in close quarters with their families, stopping and pooping near the same trails and creeks with no regard for water safety – diseases spread very fast in these conditions. From these and other stressors, about 65,000 people died in this 40-year period.
Stated another way, more people die from dysentery now, every year than ever died from any cause on the Oregon Trail. So let’s calm down about the Oregon Trail, okay?
*Lots of people use this 400,000 number but I can’t figure out where it came from and if this is referring to individuals or families – I’ve seen sources indicate it was either. If it was families, it was probably counting the men who were “the pioneers” and then being like “oh and there were women and kids there also, I guess.” But maybe it was individuals? Or maybe someone just made this up? Again, no idea where it came from. You gotta be careful every time anyone tells you a number. It’s so bad out there. The only thing worse than someone telling you a number is when they don’t tell you a number.
Getting ready
A week or so before I went, I’d been pointed to Jake Eberts’s twitter thread. Jake Eberts also participated in a challenge trial for a dysentery vaccine, also I think at UMD and the same Baltimore facility I was at, where he got very sick and went viral for livetweeting the experience. He started a fundraiser for dysentery relief and got a lot of people to sign up for clinical trials themselves, and now he works for 1DaySooner, premier “hey, human challenge trials are cool” advocates.
I read his twitter thread and sent my friends this meme:
I brought Infinite Jest, which I was partway through and was a lot more through (but still not done) by the time I was discharged. (I’m writing this while traveling, and in a fit of poor timing I finally finished it on the plane ride in, which means I now have a giant brick of a book to carry around in my suitcase.) My friend Ozy said that Infinite Jest was a really good book for reading in a dysentery ward.
I thought, oh, that’s interesting, you know, a lot of the characters are pretty miserable and living in a controlling institution of some kind. Then I remembered this one passage, where circumstances have forced a character into withdrawing from heroin alone, holed up for days in a public bathroom:
Time began to pass with sharp edges. Its passage in the dark or dim-lit stall was like time being carried by a procession of ants, a gleaming red martial column of those militaristic red Southern-U.S. ants that build hideous tall boiling hills, and each vile gleaming ant wanted a minuscule little portion of Poor Tony’s flesh in compensation as it helped bear time slowly forward down the corridor of true Withdrawal. By the second week in the stall time itself seemed the corridor, lightless at either end. After more time time then ceased to move to be moved or be move-throughable and assumed a shape above and apart, a huge, must-feathered, orange-eyed wingless fowl hunched incontinent atop the stall, with a kind of watchful but deeply uncaring personality that didn’t seem keen on Poor Tony Krause as a person at all, or to wish him well. Not one little bit. It spoke to him from atop the stall, the same things, over and over. They were unrepeatable. Nothing in even Poor Tony’s grim life-experience prepared him for the experience of time with a shape and an odor, squatting; and the worsening physical symptoms were a spree at Bonwit’s compared to time’s black assurances that the symptoms were merely hints, signposts pointing up at a larger, far more dire set of Withdrawal phenomena that hung just overhead by a string that unraveled steadily with the passage of time. It would not keep still and would not end; it changed shape and smell.
I was forced to agree that Infinite Jest was indeed probably a pretty good choice.
Two days until challenge
Checking in, everyone’s bags were checked. I got the impression they really didn’t want some kind of bad outcome where they had to call cops into a ward where everyone was running around with the bloody flux, which, fair enough. They did take away my craft scissors. I didn’t end up knitting so it wasn’t a big deal but like I’m pretty sure I’ve taken those on airplanes before. Okay.
We were assigned a number (I was just on this side of divinity at No. 107), given a plastic wristband, and shown to our rooms. We were also given two pairs of scrubs which were to be our main clothes on the ward – less risk of ruining hard-to-launder clothes in the more messy phases of the study – though it did mean 15 people having to coordinate laundry every day.
Where I made my stand
The ward was more of a retrofitted office building than a hospital. It consisted of some spaces for nurses and testing, about 6 bedrooms of various sizes (each with their own half-bath), two separate areas with two shower stalls each, a “kitchen” with snacks and where the meals were delivered to, a closet with washer and dryer, and a rec room with couches and a TV and a pool and foosball table.
There were about 16 people on the ward, an even mix of men and women. Most of them were Baltimore locals; many of them had done other trials before. We were fully allowed to socialize – dysentery is, again, infectious through the fecal-oral route, hand sanitizer was stationed all over the place but there wasn’t a huge concern that we’d infect each other or even the nurses.
Life on the ward is very chill. I was worried about being bored, but I’d forgotten that I spend most of my waking hours on the computer anyway, so it really wasn’t a problem. When even my iron gaze faltered and couldn’t stare at the computer anymore, I read Infinite Jest.
Meals were delivered once a day – one cold usually wrap- or sandwich-based meal, one hot breakfast, one hot supper dish, labelled with people’s numbers.
Sample lunch
They were, like, fine. The caterers made a few interesting choices – for vegetarians such as myself, every sandwich/wrap was some veggies with hummus, and now and then there’d be like breakfast pancakes with a curry-flavored veggie hamburger patty. I would describe the flavor when drenched with table syrup as “weird.”
Like, you can tell the person planning that menu was like “okay, pancakes and bacon… And wait, crap, something with protein for the vegetarians.” But again, I’ve eaten worse for things I’ve actually paid for ingredients for, and I was definitely eating better in terms of variety and volume than I did at home. I’m not complaining.
One day before challenge: the age of phage
This study was sort of an over-time test – ideally the first of a few, where we’d get phages before (unless we were in the control group), during, and after the “challenge” (the shigella) to see if they had any effect at all – if it did, later studies could determine if you could just drink the phage after getting sick, or if it would work best as a prophylactic, or etc. We drank a chalky buffer solution to neutralize stomach acid and give the bacteriophages (and later, the bacteria) a better chance at making it to the intestine.
What do the solutions taste like? Basically all salty fluid with slight mineral nuance, from the buffer. Phages are known to be pretty tasteless so I didn’t expect anything else.
Bacteriophage therapy: sending a cat after mice
A bacteriophage is a virus that infects bacteria. They were discovered shortly after bacteria themselves were really pinned down – microscopes were finally powerful enough to make out bacteria, and visionaries like Robert Koch and Louis Pasteur were pinpointing that these little nothing-pinpricks were in fact the source of diseases. (For more on the discovery of the microbial world, see “Through the Looking Glass and what Zheludev Et Al. (2024) Found There”, my recent piece in Asterisk Magazine.)
In 1917, Félix d’Hérelle found an agent that killed cholera bacteria, which passed through a fine filter, and which could reproduce – a living agent that killed bacteria, but that was itself smaller than a bacteria.
d’Hérelle realized right away this substance which killed bacteria, and which people had apparently been drinking, had potential as medicine. He bred pathogenic bacteria in vats and added solutions, and waited until the cloudy brother of bacteria turned clear – then offered this liquid to sick patients. Many of them, sure enough, recovered. I was (unless I was in the control group) walking in historical footsteps. Dysentery was the first human disease ever treated with phage medicine.
Sending a phage after bacteria is like sending a cat after mice. Phages are small, targeted, well-adapted hunters of specific bacteria. There is no way for them to infect a human cell like a human virus would – they are completely specialized. Phages are already in the body, along with their bacterial hosts – so you’re not introducing a radically new agent – and the immune system tends to play well with them.
Phage are used widely in some parts of the world – the Republic of Georgia and Poland both sell phage over-the-counter, for use in say intestinal conditions or wounds, and have clinics for personalized treatment. In the US, phage therapy is an extremely rare specialty, sometimes even falling under the umbrella of naturopathy. (A phage being a natural bioactive product.)
Why would you use antibiotics instead of phages, or vice versa?
Phages
Antibiotics
Targeted – a phage attacks one species or one strain of bacteria Easy to find usable new ones More finnicky (e.g. less stable) Predator-prey pharmacokinetics
Mostly spread where the bacteria are Very few side effects
Broad-spectrum
Hard to find usable new ones Shelf stable Regular blood-elimination-curve pharmacokinetics Systemic; enter the bloodstream Sometimes-serious side effects
What if the bacteria become resistant to the phages too?
Well, that can happen easily – probably even easier than with antibiotics. Cells have been duking it out with viruses since the beginning of life. (Did you know CRISPR-Cas9, now used for gene editing, evolved in nature as a way for bacteria to recognize and cut up phage DNA?)
But the difference is that whereas new antibiotics are very hard to find, there is a nigh-inexhaustible evolutionary font of phages constantly pulling ahead in the arms race. So in short: once a bacteria becomes resistant to your special phage, just find a new phage.
Do they work?
To my knowledge, there aren’t any really gold-standard reviews comparing phages head-on to antibiotics. They are fiddlier than antibiotics, with a specialized body of knowledge for treatment – less stable, have to be introduced to the site directly, much more care in choosing an appropriate treatment.
One small study found a phage treatment comparably effective to antibiotics for Salmonella typhimirium in 36 lab mice.1 Another meta-study compared modern antibiotic studies to 17 studies from the last time human phage research was in vogue in the US, the 1920s-40s, and found that phages were effective treatments – but 4 modern clinical trials suggested phages were not effective.2 A more recent study of personalized phage therapy showed promising results in infections considered “difficult-to-treat”.3 They seem to work best when used with antibiotics.
I’m not doing a full lit review right now. I bet that phage therapy still has promise – more careful formulations and just more research will help. That’s before challenges of commercial rollout, including things like handling FDA approval for a product that must be reformulated regularly.
The elephant in the room is antibiotic resistance – antibiotics usually work extremely well, but increasingly, bacteria can survive them. Antibiotic resistance is, unlike other diseases you might think of that are exacerbated by over-medication, not a condition of privileged countries – lots of Shigella bacteria in developing countries are increasingly antibiotic-resistant.
Even if phages don’t work as well as the magic silver bullet that is antibiotics, they might work well enough to be worth incorporating into our medical toolbox as part of AMR management. And that means developing them now.
The other challenge is of course regulatory – I’m excited that Intralytix, who made the experimental product I did-or-didn’t take, is throwing their hat into the space of human phage medicine, and to see how they handle this.
Day 1 of challenge
On the third day in the ward after a day of baseline and a day of phage (unless we were in the control group), we took another dose of phage (unless we were in the control group), waited a couple of hours, and then drank a glass of shigella. This tasted like baking soda and salt with no particular nuance, nor would I expect nuance; the dose was some 1300 organisms – as in 1300 individual cells of bacteria, count ‘em. A preposterously scant microbial innoculum, even for devoted parasites it often takes on the order of millions of organisms to lodge an infection – but shigella is remarkably tenacious. It would only have taken 10-200. This was overkill – a dose that WILL make you sick, unless you’re protected. All the participants drank.
The waiting game
Shigella has a 24-72 hour incubation period, maybe 12-96 hours on the far ends.
Perhaps owing to quirks of my own psyche, whose origins I’m sure we don’t need to explore here, I find it reassuring to have reference experiences to conveniently benchmark the rest of my life by. If you go skiing, you can ask yourself later, “is this more or less exhilarating than skiing?” If you fall in love once, you can compare future loves to that earlier experience.
A good standard reference point for “shared, resigned dread” is the 72 or so hours in a clinical trial ward after everyone has ingested shigella bacteria along with maybe-a-treatment.
The vibes were ominous. Jovially nervous. Unprecedented gastrointestinal distress may or may not have been coming for me, but if it is, it would be arriving in (on average) 48 hours.
The floor was pretty quiet. The hours ticked by.
Infinite Jest is, by the way, a great book. David Foster Wallace knew how to write a goddamn sentence on purpose.
Let’s learn about Shigella pathogenesis
While I waited, I decided to read up. Shigella bacteria invades the body via the digestive canal, and infects the intestines – both small and large. It releases a toxin that facilitates its infection of other parts of the intestine and its eventual replication. It’s an intracellular pathogen – some bacteria, like all viruses, actually enter the host’s cell and replicate inside there.
Shigella actually prefers to invade the outside (or should I say the inside?) of intestinal cells. But the body is a locked-down system with its own guard force, the immune system, keeping the dirty external environment separate from the sterile inside environment. Shigella in the digestive tract really wants to poke through that line of intestinal cells and get at them from the other side.
Once inside of the colon, S. flexneri can penetrate the epithelium in three ways: 1) The bacterium can alter the tight junctions between the epithelial cells, allowing it to cross into the sub-mucosa. 2) It can penetrate the highly endocytic M cells that are dispersed in the epithelial layer and cross into the sub-mucosa. 3) After reaching the sub-mucosa, the bacteria can be phagocytosed by macrophages and induce apoptosis, cell death. This releases cytokines that recruit polymorphonuclear cells (PMN) to the sub-mucosa. S. flexneri still in the lumen of the colon traverse the epithelial lining as the PMNs cross into the infected area. The influx of PMN cells across the epithelial layer in response to Shigella disrupts the integrity of the epithelium allowing lumenal bacteria to cross into the sub-mucosa in an M-cell independent mechanism.
This is really funny. Okay, imagine there’s a blockade of tightly parked police cars facing you and you and your buddies need to go get to their trunks so you can hide in them. Here are 3 ways to do this:
Push the police cars to the side so you can walk between them
Look for the police cars with the biggest doors, so that you can squeeze through the car and leave through their trunk (or I guess probably just stay in the trunk at that point)
Get yourself and your buddies arrested, then when they send backup police vans to push through the police to arrest all of you, run through the cracks in the blockade that those vans open up. Then go to the trunks of the original cop cars.
And then once you’re inside the car, you can open the doors between the cop cars (they’re sliding doors) and then travel laterally between the cop cars. I love cells.
As a fun side note, Shigella – including the strain I was developing an intimate relationship with, Shigella flexneri – is, taxonomically speaking, a kind of Escherichia coli. Now you may notice from the scientific nomenclature that this is not how this is supposed to work.
When genotyping was developed and applied to some familiar standby kinds of bacteria that microbiology-as-science figured it understood pretty well, researchers learned two surprising new things:
E. coli is not a coherent species. Different strains of E. coli – known to have slightly different properties, but thought to be all slight variations on the same basic species – turned out to have only 20% of their genes in common. (Humans and our closest relatives, chimpanzees, have almost all of our genes in common* and still aren’t considered as the same genus.)
Shigella is in that umbrella of shared genes – a secret family member known as a taxon in disguise. It’s more similar to many E. colis than some E. colis.
For most species, the procedure at this point would be to throw in the towel and reclassify – Escherichia coli spp. shigella, perhaps. But in this case, shigatoxin-producing Shigella and other pathogenic Escherichia coli have different enough clinical presentations that the distinction is still medically valuable, so accurate nomenclature has bowed its head to practicality. Cool! (Compare and contrast with trees.)
*Wait, don’t people talk about 99% or something? That number is actually about sequence similarityand not related genes – if we have 96% sequence similarity, meaning the exact same genetic code, probably even more of that genome is still in related genes. Genes can code for clearly related proteins/sequences and still not be identical, like they came from a common ancestor and haven’t diverged much but have picked up a few changes along the way. Different E. coli have 80% completely different genes – a human has maaaybe 50 genes that a chimp doesn’t? I didn’t try very hard to find the actual similar metric between them. It’s what I was telling you about numbers. You gotta watch out.
Let’s really learn about Shigella pathogenesis
Some 24 hours in, the first people started going down. Via word of mouth I heard the phrase “Exorcist-style projectile vomiting” used to describe someone in the next room over, a description whose accuracy I fortunately cannot verify. Most people were in their rooms all day anyhow, but the crowd in the kitchen at mealtimes or showing up for morning dosing got thinner.
I really held out. Going to bed at end of the second night, I felt okay, but couldn’t sleep well – nerves, I thought, or the faint distorted unpleasant bodily noises from other parts of the ward. I maybe managed a couple hours of sleep by the wee hours.
48 hours in, I woke up for vitals and dosing at 6 AM and started feeling really faint on the short walk to the next room. I stumbled over to the toilet. Off to the races!
I should be clear in this section that I was in as close to zero long-term danger as you can get with dysentery, which is damn close – this was in a controlled setting with doctors and nurses, monitoring my condition regularly, with a known pathogen with a known cure. In this case, we weren’t expected to languish in indefinite misery – they wanted to see if we got sick and then how sick we got, yes, but only up to a point, at which point they would “call it” – administer regular antibiotics and end our experimental treatment.
All I had to do was let the time pass.
The next few hours were very bad. Surprisingly, the gastrointestinal symptoms were not much of a problem for me – I had them, but it wasn’t much worse than those of regular food poisoning. I didn’t throw up. I just wanted to go back to sleep.
But sleep wasn’t coming.
First was the plague of chills. The institutional cotton blankets did nothing; four of them also did nothing, as if there was no heat to hold in. Freezing, tooth-clattering cold.
Within an hour came the plague of joint pain. It sank in rather quickly and was all in the lower extremities – hips, legs. Any more than one blanket became too heavy to bear having on them, so off they go, freezing cold but they weren’t palpably doing anything anyway. Right? I remembered reading people with chronic pain reporting that sometimes laying down was worse than other positions, and sure enough sitting up was – somehow – mildly better. I situated the adjustable bedside table so that I could slump onto it and maybe even sleep like that, but sleep remained out of reach.
Time wasn’t shitting so much as dragging, by the bones, over rough pavement, every second another six inches, grating, relentless, second after second after second. Time is space in which you are moved forward one way or another. Pain is an active process.
Around three hours later, the doctor came in and judged that I was done – they were calling it – symptomatically I had reached the Clinical Endpoint and would be treated. I was handed tylenol and antibiotics.
I’d always thought of tylenol as sort of a second-rate painkiller, probably worth trying if you couldn’t find ibuprofen, but damn if that tylenol didn’t work pretty quickly. As soon as I could I went to sleep for like four hours – which, as usual, if you are in a position of needing four hours of sleep, makes a lot of things better and more manageable once you can swing it.
Out the other side
The antibiotics worked really quickly. Within hours, the fever had vanished and the aches had dwindled to twinges. Within a couple days, even the gastrointestinal situation was back to normal. Other people were harder hit, other people were just starting to get sick – staying vanished in their rooms even after I stuck my head into the kitchen and rec rooms like the first hopeful groundhog of spring – and many had been fine the whole time.
The thing that kills people in dysentery is dehydration and complications thereof. So part of the recovery is collecting and measuring how many fluids were emitted, and then re-administering oral rehydration fluid – a salty liquid served ice-cold – in precise ratios to replace the bodily fluids lost. A human is a series of tubes with attached nervous system and fortunately I was in the company of master plumbers. Once the diarrhea had stopped, I was also able to stop guzzling big plastic cups of what I liked to imagine tasted like arctic seawater. Progress!
Great view from the rec room.
People who recovered and who never got sick started hanging out in the rec room more, chatting and playing pool. I spent my birthday calling my parents and talking to internet friends. One streamed himself playing a fish-themed video game in my honor. The Baltimoreans inexplicably set off fireworks many nights – maybe the proximity to July 4th? – and this was one of them. Not roadside-stand-ground fireworks, but big aerial fireworks. A fellow subject found ice cream bars in the kitchen freezer and kindly brought me one as a present. Fireworks aside, it was a quiet day.
Apologies for the deception, reader. Technically speaking, the word “dysentery” usually refers to a syndrome, like “psychosis” or “high blood pressure”, which can have multiple causes but which is defined by specific symptoms. The specific symptom of dysentery is bloody diarrhea. I personally did not get this particular symptom – I became sick with shigellosis but, according to a common criteria, did not get dysentery. I’m sorry for clickbaiting you. In my defense, I would have taken it over the joint pain.
Aftermath
Twice a day after antibiotics, we gave the nurses a stool sample – these were sampled and cultured at some lab to determine if shigella was still in there. Two negative samples in a row meant that we were free to go.
9 days after coming in, I was cleared for release. I collected my scissors, and, free of dysentery, was released onto the streets of Baltimore. A year older on paper. Healthy, wrung out, ready for time to keep doing what it does. Hopefully, mostly on kinder terms.
The train ride home. I see that 75% of these photos have coffee in them. What can I say? I’m from Seattle.
I think that despite my relatively mild case, that I was in the control group. But the reason I think that was because in the whole trial, everyone drank the shigella, and it sure seemed like about half of them didn’t get sick at all.
Luigi Marongiu et al., “Reassessment of Historical Clinical Trials Supports the Effectiveness of Phage Therapy,” Clinical Microbiology Reviews 35, no. 4 (September 7, 2022): e00062, https://doi.org/10.1128/cmr.00062-22. ↩︎
Jean-Paul Pirnay et al., “Personalized Bacteriophage Therapy Outcomes for 100 Consecutive Cases: A Multicentre, Multinational, Retrospective Observational Study,” Nature Microbiology 9, no. 6 (June 2024): 1434–53, https://doi.org/10.1038/s41564-024-01705-x. ↩︎
Thank you Grace Neptune, Kelardry, and YumAntimatter for reviewing a draft of this post.
I have a Patreon! Consider supporting my writing by throwing me a few bucks. I’d really appreciate it. I won’t be getting dysentery again (…on purpose) but I have some other good stuff in the works.
I’ve written for Asterisk before: What I won’t eat, on arriving at an equilibrium on the “it’s bad when animals suffer” vs. “but animal products taste good” challenge.
A novel lethal infectious neurological disease emerged in American deer a few decades ago. Since then, it’s spread rapidly across the continent. In areas where the disease is found, it can be very common in the deer there.
Chronic wasting disease isn’t caused by a bacteria, virus, protist, or worm – it’s a prion, which is a little misshapen version of a protein that occurs naturally in the nervous systems of deer.
Chemically, the prion is made of exactly the same stuff as its regular counterpart – it’s a string of the same amino acids in the same order, just shaped a little differently. Both the prion and its regular version (PrP) are monomers, single units that naturally stack on top of each other or very similar proteins. The prion’s trick is that as other PrP moves to stack atop it, the prion reshapes them – just a little – so that they also become prions. These chains of prions are quite stable, and, over time, they form long, persistent clusters in the tissue of their victims.
We know of only a few prion diseases in humans. They’re caused by random chance misfolds, a genetic predisposition for PrP to misfold into a prion, accidental cross-contamination via medical supplies, or, rarely, from the consumption of prion-infected meat. Every known animal prions is a misfold of the same specific protein, PrP. PrP is expressed in the nervous system, particularly in the brain – so infections cause neurological symptoms and physical changes to the structure of the brain. Prion diseases are slow to develop (up to decades), incurable, and always fatal.
There are two known infectious prion diseases in people. One is kuru, which caused an epidemic among tribes who practiced funerary cannibalism in Papua New Guinea. The other is mad cow disease, also known as bovine spongiform encephalopathy (BSE) AKA Variant Creutzfeldt-Jakob disease, which was first seen in humans in 1996 in the UK, and comes from cows.
Chronic wasting disease (CWD)…
Is, like every other animal prion disease, a misfold of PrP. PrP is quite similar in both humans and deer.
Is found in multiple deer species which are commonly eaten by humans.
Can be carried in deer asymptomatically.
But it doesn’t seem to infect people. Is it ever going to? If a newly-emerged virus were sweeping across the US and killing deer, which could be spread through consuming infected meat, I would think “oh NO.” I’d need to see very good evidence to stop sounding the alarm.
Now, the fact that it’s been a few decades, and it hasn’t spread to humans yet, is definitely some kind of evidence about safety. But are we humans basically safe from it, or are we living on borrowed time? If you live in an area where CWD has been detected, should you eat the deer?
Sidenote: Usually, you’ll see “BSE” used for the disease in cows and “VCJ” for the disease in humans. But they’re caused by the same agent and this essay is operating under a zoonoticOne Health kind of stance, so I’m just calling the disease BSE here. (As well as the prion that causes it, when I can get away with it.)
In short
The current version of CWD is not infectious to people. We checked. BSE showed that prions can spill over, and there’s no reason a new CWD variant will never do the same. The more cases there are, the more likely it is to spill over. That said, BSE did not spill over very effectively. It was always incredibly rare in humans. It’s an awful disease to get, but the chance of getting it is tiny. Prions in general have a harder time spilling over between species than viruses do. CWD might behave somewhat differently but probably will stay hampered by the species barrier.
Why do I think all of this? Keep reading.
North American elk (wapiti), which can carry CWD. This and the image at the top of the article are adapted from a photo from the Idaho Fish and Game department, under a CC BY 2.0 license.
Prions aren’t viruses
I said before that if a fatal neurological virus were infecting deer across the US, and showed up in cooked infected meat, my default assumption would be “we’re in danger.” But a prion isn’t a virus. Why does that matter?
Let’s look at how they replicate. A virus is a little bit of genetic material in a protein coating. You, a human, are a lot of genetic in a protein coating. When a virus replicates, it slips into your cells, and it hijacks your replication machinery to run its genes instead. Instead of all the useful-to-you tasks your genome has planned, the virus’s genome outlines its own replication, assembles a bunch more viruses, and blows up the factory (cell) to turn them loose into the world.
In other words, the virus using a robust information-handling system that both you and it have in common – the DNA → RNA → protein pipeline often called “the central dogma” of biology. To a first approximation, you can just add any genetic information at all into the viral genome, and as long as it doesn’t interfere with the virus’s process, whatever you add will get replicated in there too.
Prions do not work like this. They don’t tap into the central dogma. What makes them so fundamentally cool is that they replicate without touching the replication machinery that everything else alive uses – their replication is structural, like a snowflake forming. The host provides raw material in the form of PrP, and the prion – once it lands – encourages that material to shape in the right way for more to form atop it.
What this means is that you can’t encode arbitrary information into a prion. This isn’t just a factor – it’s not as though a prion runs on a separate “protein genome” we could decipher and then encode what we like into. The entire structure of the prion has to work together to replicate itself. If you made a prion with some different fold in it, that fold has to not just form a stable protein, but to pass itself along as well. They don’t have a handy DNA replicase enzyme to outsource to – they have to solve the problem of replication themselves, every time.
Prions can evolve, but they do it less – they have fewer free options, they’re more constrained than a virus would be in terms of changes that don’t interrupt the rest of the refolding process and that on top of that promulgate themselves.
This means that prions are slower to evolve than viruses. …I’m pretty sure, at least. It makes a lot of sense to me. The thing that this definitely means is that:
It’s very hard for prions to cross species barriers
PrP is a very conserved protein across mammals, meaning that all mammals have a version of PrP that’s pretty similar – 90%+ similarity.* But the devil lies in that 10%.
Prions are finely tuned – to convert PrP to a prion, it basically needs to be identical, or at least functionally identical, everywhere the prion works. It not just needs to be susceptible to the prion’s misfolding, it also needs to fold into something that itself can replicate. A few amino acid differences can throw a wrench in the works.
It’s clear that infectious prions can have a hard time crossing species barriers. It depends on the strain. For instance: Mouse prions convert hamster PrP.** Hamster prions don’t convert mouse PrP. Usually a prion strain converts its usual host PrP best, but one cat prion more efficiently converts cow PrP. In a test tube, CWD can convert human or cow PrP a little, but shows slightly more action with sheep PrP (and much more with, of course, deer PrP.)
This sounds terribly arbitrary. But remember, prion behavior comes down to shape. Imagine you’re playing with legos and duplo blocks. You can stack legos on legos and duplos on duplos. You can also put a duplo on top of a lego block. But then you can only add duplo blocks on top of that – you’ve permanently changed what can get added to that stack.
When we look at people – or deer, or sheep, etc – who are genetically resistant to prions (more on that later), we find that serious resistance can be conferred by single nucleic acid changes in the PrP gene. Tweak one single letter of DNA in the right place, and their PrP just doesn’t bend into the prion shape easily. If the infection takes, it proceeds slower– slow enough a person might die of old age before the prion would kill them.
So if a decent number of members of a species can be resistant to prion diseases, based on as little as one amino acid – then a new species, one that might have dozens of different amino acids in the PrP gene, is unlikely to be fertile ground for an old prion.
* (This is kind of weird given that we don’t know what PrP actually does – the name PrP just stands “prion protein” because it’s the protein that’s associated with prions, and we don’t know its function. We can genetically alter mice so that they don’t produce PrP at all, and they show slight cognitive issues but they’re basically fine. Classic evolution. It’s appendices all over again.)
** Sidebar: When we look at studies for this, we see that like a lot of pathology research, there's a spectrum of experiments on different points on the axis from “deeply unrealistic” to “a pretty reasonable simulacrum of natural infection”, like:
1. Shaking up loose prions and PrP in a petri dish and seeing if the PrP converts
2. Intracranial injection with brain matter (i.e. grinding up a diseased brain and injecting some of that nasty juice into the brain of a healthy animal and seeing if it gets sick)
3. Feeding (or some other natural route of exposure) a plausible natural dose of prions to a healthy animal and seeing if that animal gets sick
The experiments mentioned below are based on 1. Only experiments that do 3 actually prove the disease is naturally infectious. For instance, Alzheimer’s disease is “infectious” if you do 2, but since nobody does that, it’s not actually a contagious threat. That said, doing more-abstracted experiments means you can really zoom in on what makes strain specificity tick.
But prions do cross species barriers
Probably the best counterargument to everything above is that another prion disease, BSE, did cross the species barrier. This prion pulled off a balancing act: it successfully infected cows and humans at the same time.
Let’s be clear about one big and interesting thing: BSE is not good at crossing the species barrier. When I say this, I mean two things:
First, people did not get it often. While the big UK outbreak was famously terrifying, only around 200 people ever got sick from mad cow disease. Around 200,000 cows tested positive for it. But most cows weren’t tested. Researchers estimate that 2 million cows total in the UK had BSE, most of which were slaughtered and entered the food chain. These days, Britain has 2 million cows at any given time.
At first glance, and to a first approximation, I think everyone living in the UK for a while between 1985 and 1996 or so (who ate beef sometimes) must have eaten beef from an infected animal. That’s approximately who the recently-overturned blood donation ban in the US affected. I had thought that was sort of an average over who was at risk of exposure – but no, that basically encompassed everyone who was exposed. Exposure rarely leads to infection.
You’re more likely to get struck by lightning than to get BSE even if you have eaten BSE-infected beef.
Second, in the rare cases the disease takes,it’s slower. Farm cows live short lives, and the cows that died from BSE would have gotten old for the beef industry at 4-5 years post-exposure. They survived at most weeks or months after symptoms began. Humans infected with BSE, meanwhile, can harbor it for up to decades post-exposure, and live an average of over a year after showing symptoms.
I think both of these are directly attributable to the prion just being less efficient at converting human PrP – versus the PrP of the cows it was adapted to. It doesn’t often catch on in the brain. When it does, it moves extremely slowly.
But it did cross over. And as far as I can tell, there’s no reason CWD can’t do the same. Like viruses, CWD has been observed to evolve as it bounces between hosts with different genotypes. Some variants of CWD seem more capable of converting mouse PrP than the common ones. The good old friend of those who play god, serial passaging, encourages it.
(Note also that all of the above differs from kuru, which did cause a proper epidemic. Kuru spread between humans and was adapted for spreading in humans. When looking to CWD, BSE is a better reference point because it spread between cows and only incidentally jumped to humans – it was never adapted for human spread.)
How is CWD different from BSE?
BSE appears in very low, very low numbers anywhere outside the brains and spines of its victims. CWD is also concentrated in the brains, but also appears in the spines and lymphatic tissue, and to a lesser but still-present degree, everywhere else: muscle, antler velvet, feces, blood, saliva. It’s more systematic than BSE.
Cows are concentrated in farms, and so are some deer, but wild deer carry CWD all hither and yon. As they do, they leave it behind in:
Bodies – Deer aren’t strictly herbivorous if push comes to shove. If a deer dies, another deer might eat the body. One study found that after a population of reindeer started regularly gnawing on each other’s antlers (#JustDeerThings), CWD swept in.
Dirt – Prions are resilient and can linger, viable, in soil. Deer eat dirt accidentally while eating grass, as well as on purpose from time to time and can be infected.
Grass – Prions in the soil or otherwise deposited onto plant tissue can hang out in living grass for a long time.
Ticks – One study found that ticks fed CWD prions don’t degrade the protein. If they’re then eaten by deer (for instance, during grooming), they could spread CWD. This study isn’t perfect evidence; the authors note that they fed the ticks a concentration of prions about 1000x higher than is found in infected deer blood. But if my understanding of statistics and infection dynamics is correct, that suggests that maybe 1 in 1000 ticks feeding on infected deer blood reaches that level of infectivity? Deer have a lot of ticks! Still pretty bad!
That’s a lot of widespread potentially-infectious material.
When CWD is in an area, it can be very common – up to 30% of wild deer, and up to 90% of deer on an infected farm. These deer can carry CWD and have it in their tissues for quite some time asymptomatically – so while it frequently has very visible behavioral and physical symptoms, it also sometimes doesn’t.
In short, there’s a lot of CWD in lots of places through the environment. It’s also spreading very rapidly. If a variant capable of infecting both deer and humans emerged, there would be a lot of chances for possible exposure.
As with any circumstance at all, COVID or salmonella or just living in a world that is sometimes out to get you, you have to choose what level of risk you’re alright with. At first, writing this piece, I was going to make a suggestion like “definitely avoid eating deer from areas that have CWD just in case your deer is the one that has a human-transmissible prion disease.” I made a little chart about my sense of the relative risk levels, to help put the risk in scale even though it wasn’t quantified. It went like this:
But, as usual, quantification turns out to be pretty important. I actually did the numbers about how many people ever got sick from BSE (~200) and how many BSE-infected cows were in the food chain (~2,000,000), which made the actual risk clear. So I guess the more prosaic version looks like this:
…This is sort of a joke, to be clear. There’s not a health agency anywhere on earth that will advise you to eat meat from cows known to have BSE, and the CDC recommends not eating meat from deer that test positive for CWD (though it’s never infected a human before.)
On top of that, the overall threat is still uncertain because what you’re betting on is “the chance that this animal will have had an as-of-yet undetected CWD variant that can infect humans.” There’s inherently no baseline for that!
We don’t know what CWD would act like if it spilled over. It might be more infectious and dangerous than other infectious prion diseases we’ve seen – remember, with humans, the sample size is 2! So if CWD is in your area and it’s not a hardship to avoid eating deer, you might want to steer clear. …But the odds are in your favor.
As a society
There’s not an obvious solution. The epidemic spreading among deer isn’t caused by a political problem, it’s from nature.
The US is doing a lot right: mainly, it is monitoring and tracking the spread of the disease. It’s spreading the word. (If nothing else, you can keep track of this by subscribing to google alerts for “chronic wasting disease”, and then pretty often you’ll get an email saying things like “CWD found in Florida for the first time” or “CWD found an hour from you for the first time.”) It is encouraging people to submit deer heads for testing, and not to eat meat from deer that test positive. The CDC, APHIS, Fish & Wildlife Service, and more are all aware of the problem and participating in tracking it.
What more could be done? Well, a lot of the things that would help a potential spillover of CWD look like actions that can be taken in advance of any threatening novel disease. There is research being done on prions and how they cause disease, better diagnostics, and possible therapeutics. All of these are important. Prion disease diagnosis and treatment is inherently difficult, and on top of that, has little overlap with most kinds of diagnosis or treatment. It’s also such a rare set of diseases that it’s not terribly well studied. (My understanding is that right now there are various kinds of tests for specific prion diseases – which could be adapted for a new prion disease – that are extremely sensitive although not particularly cheap or widespread.)
I don’t know a lot about the regulatory or surveillance situation vis-a-vis deer farms, or for that matter, much about deer farms at all. I do know that they seem to be associated with outbreaks, and heavy disease prevalence once there is an outbreak. That’s a smart area to an eye on.
If CWD did spill over, what would happens?
It will probably also take time to locate cases and identify the culprit, but given the aforementioned awareness and surveillance of the issue, it ought to take way less time than it took to identify the causative agent of BSE. Officials are already paying attention to deaths that could potentially be CWD-related, like neurodegenerative illnesses that kill young people.
First, everyone gets very nervous about eating venison for a while.
After that, I expect the effects will look a lot like the aftermath of mad cow disease. Mad cow disease, and very likely a hypothetical CWD spillover, would not be transmissible between people in usual ways – coughing, skin contact, fomites, whatever.
It is transmissible via unnatural routes, which is to say, blood transfusions. You might remember how people who’d spent over 6 months in Britain couldn’t donate blood in the US until 2022, a direct response to the BSE outbreak. Yes, the disease was extremely rare, but unless you can quickly and cheaply test incoming blood donations, a donor could donate blood to multiple people. Suppose some of them donate blood down the line. You’d have a chain of infection and a disease with a potentially decades-long incubation period. And remember, the disease is incurable and fatal. So basically, the blood donation system (and probably other organ donation) becomes very problematic.
That said, I don’t think it would break down completely. In the BSE case, lots of people in the UK eat beef from time to time – probably most people. But with a deerborne disease, I would guess that a lot of the US population could confidently declare that they haven’t eaten deer within the past, say, year or so (prior to a detected outbreak.) So I think there’d be panic and perhaps strain on the system but not necessarily a complete breakdown. Again, all of this is predicated on a new prion disease working like known human prion diseases.
Genetic resistance
One final fun fact: People who have a certain allele in the PrP gene – specifically, have the genotype PRNP 129M/V or V/V – are incredibly genetically resistant to known infectious prion diseases. If they do get infected, they survive for much longer.
It’s also not clear that this would hold true for a hypothetical CWD crossover to humans. But it is true for both kuru and BSE. It’s also partly (although not totally) protective against sporadic Creutzfeldt-Jakob disease.
If you’ve gotten a service like 23&me, maybe check out your data and see if you’re resistant to infectious prion diseases. Here’s what you’re looking for:
129M/V or V/V (amino acids), or G/G or A/G (nucleotides) – rs1799990
If you instead have M/M (amino acids) or A/A (nucleotides) at that site, you’re SOL at a higher but still very low overall risk.
Final thoughts
I think exercises like “if XYZ disease emerges, what will the ramifications and response be” are valuable. They lead to questions like “what problems will seem obvious in retrospect” and “how can we build systems now that will improve outcomes of disasters.” This is an interesting case study and I might revisit it later.
Has anyone reading this ever been struck by lightning? That’s the go-to comparison for things being rare. But 1 in 15,000 isn’t, like, unthinkably rare. I’m just curious.
No, seriously, what’s the deal with deer farms? I never think about deer farms much. When I think of venison, I imagine someone wearing camo and carrying a rifle out into a national forest or a buddy’s backyard or something. How many deer are harvest from hunting vs. farms? What about in the US vs. worldwide? Does anyone know? Tell me in the comments.
If you want to encourage my work, check out my Patreon. Today’s my birthday! I sure would appreciate your support.
Also, this eukaryote is job-hunting. If you have or know of a full-time position for a researcher, analyst, and communicator with a Master’s in Biodefense, let me know:
Eukaryote Writes Blog (at) gmail (dot) com
In the mean time, perhaps you have other desires. You’d like a one-off research project, or there’s a burning question you’d love a well-cited answer to. Maybe you want someone to fact-check or punch up your work. Either way, you’d like to buy a few hours of my time. Well, I have hours, and the getting is good. Hit me up! Let’s chat. 🐟
So you’ve heard about how fish aren’t a monophyletic group? You’ve heard about carcinization, the process by which ocean arthropods convergently evolve into crabs? You say you get it now? Sit down. Sit down. Shut up. Listen. You don’t know nothing yet.
“Trees” are not a coherent phylogenetic category. On the evolutionary tree of plants, trees are regularly interspersed with things that are absolutely, 100% not trees. This means that, for instance, either:
The common ancestor of a maple and a mulberry tree was not a tree.
The common ancestor of a stinging nettle and a strawberry plant was a tree.
And this is true for most trees or non-trees that you can think of.
I thought I had a pretty good guess at this, but the situation is far worse than I could have imagined.
CLICK TO EXPAND. Partial phylogenetic tree of various plants. TL;DR: Tan is definitely, 100% trees. Yellow is tree-like. Green is 100% not a tree. Sourced mostly from Wikipedia.
I learned after making this chart that tree ferns exist (h/t seebs), which I think just emphasizes my point further. Also, h/t kithpendragon on LW for suggestions on increasing accessibility of the graph.
Why do trees keep happening?
First, what is a tree? It’s a big long-lived self-supporting plant with leaves and wood.
Also of interest to us are the non-tree “woody plants”, like lianas (thick woody vines) and shrubs. They’re not trees, but at least to me, it’s relatively apparent how a tree could evolve into a shrub, or vice-versa. The confusing part is a tree evolving into a dandelion. (Or vice-versa.)
Wood, as you may have guessed by now, is also not a clear phyletic category. But it’s a reasonable category – a lignin-dense structure, usually that grows from the exterior and that forms a pretty readily identifiable material when separated from the tree. (…Okay, not the most explainable, but you know wood? You know when you hold something in your hand, and it’s made of wood, and you can tell that? Yeah, that thing.)
All plants have lignin and cellulose as structural elements – wood is plant matter that is dense with both of these.
Botanists don’t seem to think it only could have gone one way – for instance, the common ancestor of flowering plants is theorized to have been woody. But we also have pretty clear evidence of recent evolution of woodiness – say, a new plant arrives on a relatively barren island, and some of the offspring of that plant becomes treelike. Of plants native to the Canary Islands, wood independently evolved at least 38 times!
One relevant factor is that all woody plants do, in a sense, begin life as herbaceous plants – by and large, a tree sprout shares a lot of properties with any herbaceous plant. Indeed, botanists call this kind of fleshy, soft growth from the center that elongates a plant “primary growth”, and the later growth from towards the outside which causes a plant to thicken is “secondary growth.” In a woody plant, secondary growth also means growing wood and bark – but other plants sometimes do secondary growth as well, like potatoes in their roots.
This paper addresses the question. I don’t understand a lot of the closely genetic details, but my impression of its thesis is that: Analysis of convergently-evolved woody plants show that the genes for secondary woody growth are similar to primary growth in plants that don’t do any secondary growth – even in unrelated plants. And woody growth is an adaption of secondary growth. To abstract a little more, there is a common and useful structure in herbaceous plants that, when slightly tweaked, “dendronizes” them into woody plants.
Dendronization – Evolving into a tree-like morphology. (In the style of “carcinization“.) From ‘dendro‘, the ancient Greek root for tree.
Can this be tested? Yep – knock out a couple of genes that control flower development and change the light levels to mimic summer, and researchers found thatArabidopsis – rock cress, a distinctly herbaceous plant used as a model organism – grows a woody stem never otherwise seen in the species.
So not only can wood develop relatively easily in an herbal plant, it can come from messing with some of the genes that regulate annual behavior – an herby plant’s usual lifecycle of reproducing in warm weather, dying off in cool weather. So that gets us two properties of trees at once: woodiness, and being long-lived. It’s still a far cry from turning a plant into a tree, but also, it’s really not that far.
“Obviously, in the search for which genes make a tree versus a herbaceous plant, it would be folly to look for genes present in poplar and absent in Arabidopsis. More likely, tree forms reflect differences in expression of a similar suite of genes to those found in herbaceous relatives.”
So: There are no unique “tree” genes. It’s just a different expression of genes that plants already use. Analogously, you can make a cake with flour, sugar, eggs, sugar, butter, and vanilla. You can also make frosting with sugar, butter, and vanilla – a subset of the ingredients you already have, but in different ratios and use.
But again, the reverse also happens – a tree needs to do both primary and secondary growth, so it’s relatively easy for a tree lineage to drop the “secondary” growth stage and remain an herb for its whole lifespan, thus “poaizating.” As stated above, it’s hypothesized that the earliest angiosperms were woody, some of which would have lost that in become the most familiar herbaceous plants today. There are also some plants like cassytha and mistletoe, herbaceous plants from tree-heavy lineages, who are both parasitic plants that grow on a host tree. Knowing absolutely nothing about the evolution of these lineages, I think it’s reasonable to speculate that they each came from a tree-like ancestor but poaized to become parasites. (Evolution is very fond of parasites.)
Poaization: Evolving into an herbaceous morphology. From ‘poai‘, ancient Greek term from Theophrastus defining herbaceous plants (“Theophrastus on Herbals and Herbal Remedies”).
(I apologize to anyone I’ve ever complained to about jargon proliferation in rationalist-diaspora blog posts.)
The trend of staying in an earlier stage of development is also called neotenizing. Axolotls are an example in animals – they resemble the juvenile stages of the closely-related tiger salamander. Did you know very rarely, or when exposed to hormone-affecting substances, axolotls “grow up” into something that looks a lot like a tiger salamander? Not unlike the gene-altered Arabidopsis.
A normal axolotl (left) vs. a spontaneously-metamorphosed “adult” axolotl (right.)
A friend asked why I was so interested in this finding about trees evolving convergently. To me, it’s that a tree is such a familiar, everyday thing. You know birds? Imagine if actually there were amphibian birds and mammal birds and insect birds flying all around, and they all looked pretty much the same – feathers, beaks, little claw feet, the lot. You had to be a real bird expert to be able to tell an insect bird from a mammal bird. Also, most people don’t know that there isn’t just one kind of “bird”. That’s what’s going on with trees.
I was also interested in culinary applications of this knowledge. You know people who get all excited about “don’t you know a tomato is a fruit?” or “a blueberry isn’t really a berry?” I was one once, it’s okay. Listen, forget all of that.
There is a kind of botanical definition of a fruit and a berry, talking about which parts of common plant anatomy and reproduction the structure in question is derived from, but they’re definitely not related to the culinary or common understandings. (An apple, arguably the most central fruit of all to many people, is not truly a botanical fruit either).
Let me be very clear here – mostly, this is not what biologists like to say. When we say a bird is a dinosaur, we mean that a bird and a T. rex share a common ancestor that had recognizably dinosaur-ish properties, and that we can generally point to some of those properties in the bird as well – feathers, bone structure, whatever. You can analogize this to similar statements you may have heard – “a whale is a mammal”, “a spider is not an insect”, “a hyena is a feline”…
But this is not what’s happening with fruit. Most “fruits” or “berries” are not descended from a common “fruit” or “berry” ancestor. Citrus fruits are all derived from a common fruit, and so are apples and pears, and plums and apricots – but an apple and an orange, or a fig and a peach, do not share a fruit ancestor.
Instead of trying to get uppity about this, may I recommend the following:
Acknowledge that all of our categories are weird and a little arbitrary
Send a fruit basket to your local botanist/plant evolutionary biologist for putting up with this, or become one yourself
While natural selection is commonly thought to simply be an ongoing process with no “goals” or “end points”, most scientists believe that life peaked at Welwitschia.
Avocado and cinnamon are from fairly closely-related tree species.
It’s possible that the last common ancestor between an apple and a peach was not even a tree.
Of special interest to my Pacific Northwest readers, the Seattle neighborhood of Magnolia is misnamed after the local madrona tree, which Europeans confused with the (similar-looking) magnolia. In reality, these two species are only very distantly related. (You can find them both on the chart to see exactly how far apart they are.)
None of [cactuses, aloe vera, jade plants, snake plants, and the succulent I grew up knowing as “hens and chicks”] are related to each other.
Rubusis the genus that contains raspberries, blackberries, dewberries, salmonberries… that kind of thing. (Remember, a genus is the category just above a species – which is kind of a made-up distinction, but suffice to say, this is a closely-related groups of plants.) Some of its members have 14 chromosomes. Some of its members have 98 chromosomes.
Seriously, I’m going to hand $20 in cash to the next plant taxonomy expert I meet in person. God knows bacteriologists and zoologists don’t have to deal with this.
And I have one more unanswered question. There doesn’t seem to be a strong tend of plants evolving into grasses, despite the fact that grasses are quite successful and seem kind of like the most anatomically simple plant there could be – root, big leaf, little flower, you’re good to go. But most grass-like plants are in the same group. Why don’t more plants evolve towards the “grass” strategy?
Let’s get personal for a moment. One of my philosophical takeaways from this project is, of course, “convergent evolution is a hell of a drug.” A second is something like “taxonomy is not automatically a great category for regular usage.” Phylogenetics are absolutely fascinating, and I do wish people understood them better, and probably “there’s no such thing as a fish” is a good meme to have around because most people do not realize that they’re genetically closer to a tuna than a tuna is to a shark – and “no such thing as a fish” invites that inquiry.
(You can, at least, say that a tree is a strategy. Wood is a strategy. Fruit is a strategy. A fish is also a strategy.)
At the same time, I have this vision in my mind of a clever person who takes this meandering essay of mine and goes around saying “did you know there’s no such thing as wood?” And they’d be kind of right.
But at the same time, insisting that “wood” is not a useful or comprehensible category would be the most fascinatingly obnoxious rhetorical move. Just the pinnacle of choosing the interestingly abstract over the practical whole. A perfect instance of missing the forest for – uh, the forest for …
Towards the end of writing this piece, I found that actual botanist Dan Ridley-Ellis made a tweet thread about this topic in 2019. See that for more like this from someone who knows what they’re talking about.
Epistemic status: Speculative, just having fun. This piece isn’t well-cited, but I can pull up sources as needed – nothing about mole-rats is my original research. A lot of this piece is based on Wikipedia.
When I wrote about “weirdness” in the past, I called marine invertebrates, archaea viruses, and Florida Man stories “predictably weird”. This means I wasn’t really surprised to learn any new wild fact about them. But there’s a sense in which marine invertebrates both are and aren’t weird. I want to try operationalizing “weirdness” as “amount of unpredictability or diversity present in a class” (or “in an individual”) compared to other members of its group.
Invertebrates represent most of the strategies that animals have attempted on earth, and certainly most of the animals on earth. Vertebrates are the odd ones out.
But you know which animals are profoundly weird, no matter which way you look at it? Naked mole rats. Naked mole-rats have like a dozen properties that are not just unusual, not just strange, but absolutely batshit. Let’s review.
1. They don’t age
What? Well, for most animals, their chance of dying goes up over time. You can look at a population and find something like this:
Mole-rats, they have the same chance of dying at any age. Their graph looks like this:
They’re joined, more or less, by a few species of jellyfish, flatworms, turtles, lobsters, and at least one fish.
They’re hugely long-lived compared to other rodents, seen in zoos at 30+ years old compared to the couple brief years that rats get.
2. They don’t get cancer
Cancer generally seems to be the curse of multicellular beings, but naked mole-rats are an exception. A couple mole-rats have developed cancer-like growths in captivity, but no wild mole-rat has ever been found with cancer.
Definitely unique among mammals. Like bees, ants, and termites, naked mole-rats have a single breeding “queen” in each colony, and other “worker” individuals exist in castes that perform specific tasks. In an evolutionary sense, this means that the “unit of selection” for the species is the queen, not any individual – the queen’s genes are the ones that get passed down.
They’re also a fascinating case study of an animal whose existence was deduced before it was proven. Nobody knew about eusocial mammals for a long time. In 1974, entomologist Richard Alexander, who studied eusocial insects, wrote down a set of environmental characteristics he thought would be required for a eusocial mammal to evolve. Around 1981 and the next decade, naked mole-rats – a perfect match for his predictions – were found to be eusocial.
5. They don’t have fur
Obviously. But aside from genetic flukes or domesticated breeds, that puts them in a small unlikely group with only some marine mammals, rhinoceros, hippos, elephants, one species of boar, and… us.
You and this entity have so much in common.
6. They’re able to survive ridiculously low oxygen levels
It uses very little oxygen during normal metabolism, much less than comparable-sized rodents, and it can survive for hours at 5% oxygen (a quarter of normal levels.)
7. Their front teeth move back and forth like chopsticks
I’m not actually sure how common this is in rodents. But it really weirded me out.
They have basically no ability to adjust their body temperature internally, perhaps because their caves tend to be rather constant temperatures. If they need to be a different temperature, they can huddle together, or move to a higher or lower level in their burrow.
All of this makes me think that mole-rats must have some underlying unusual properties which lead to all this – a “weirdness generator”, if you will.
A lot of these are connected to the fact that mole rats spend almost their entire lives underground. There are lots of burrowing animals, but “almost their entire” is pretty unusual – they don’t surface to find food, water, or (usually) mates. (I think they might only surface when digging tunnels and when a colony splits.) So this might explain (8) – no need for a sleep schedule when you can’t see the sun. It also seems to explain (5) and (9), because thermoregulation is unnecessary when they’re living in an environment that’s a pretty constant temperature.
It probably explains (6) because lower burrow levels might have very little oxygen most of the time, although there’s some debate about this – their burrows might actually be pretty well ventilated.
And Richard Alexander’s 12 postulates that would lead to a eusocial vertebrate – plus some other knowledge of eusociality – suggests that this underground climate, when combined with the available lifestyle and food source of a molerat, should lead to eusociality.
It might also be the source of (2) and (3) – people have theorized that higher CO2 or lower oxygen levels in burrows might reduce DNA damage or related to neuron function or something. (This would also explain why only mole-rats in captivity have had tumors, since they’re kept at atmospheric oxygen levels.) These still seem to be up in the air, though. Mole-rats clearly have a variety of fascinating biochemical tricks that are still being understood.
So there’s at least one “weirdness generator” that leads to all of these strange mole-rat properties. There might be more.
I’m pretty sure it’s not the chopstick teeth (7), at least – but as with many predictions one could make about mole rats, I could easily be wrong.
One thing that surprised me when working on How Many Neurons Are There was the number of neurons in the brains of very small animals.
Let’s look a classic measurement, the brain-mass:body-mass ratio.* Smarter animals generally have larger brain sizes for their body mass, compared to animals of similar size. Among large animals, humans have famously enormous brains for our size – the highest of any large animal, it seems. But as we look at smaller animals, that ratio goes up again. A mouse has a comparable brain:body-mass ratio to a human. Getting even smaller, insects have higher brain:body-mass ratios than any vertebrate we know of: more like 1 in 6.
But brain mass isn’t quite what we want – brains are mostly water, and there are a lot of non-neuron cells in brains. Conveniently, I also have a ton of numbers put together on number of neurons. (Synapse counts might be better, but those are hard to come by for different species. Ethology would also be interesting.)
And the trend is also roughly true for neuron-count:body-mass. Humans do have unusually high numbers of neurons per kilogram than other animals, but far, far fewer than, for instance, a small fish or an ant.
If you believe some variation on one of the following:
Different species have moral worth in proportion to how many neurons they have
Different animal species have moral worth in proportion to how smart they are
Different species have moral worth in proportion to the amount of complex thought they can do
Different species have moral worth in proportion to how much they can learn**
…then this explanation is an indication that insects and other small animals have much more moral worth than their small size suggests.
How much more?
Imagine, if you will, a standard 5-gallon plastic bucket.
Now imagine that bucket contains 300,000 ants – about two pounds.*** Or a kilogram, if you prefer.
Imagine the bucket. Imagine the equivalent of a couple large apples inside it.
A bucket. Two pounds of ants.
Those ants, collectively, have as many neurons as you do.
(Graphic design is my passion.)
You may notice that an adult human brain actually weighs more than two pounds. What’s going on? Simply, insect brains are marvels of miniaturization. Their brains have a panoply of space-saving tricks, and the physical cells are much smaller.
🐜🐜🐜
*Aren’t the cool kids using cephalization quotients rather than brain-mass:body-mass ratios? Yes, when it comes to measurements of higher cognition in vertebrates, cephalization is (as far as I’m aware) thought of as better. But there’s debate about that too. Referring to abilities directly probably makes sense for assessing abilities. I don’t know much about this and it’s not the focus of this piece, anyway.
**Yes, I know that only the first question is directly relevant to this piece, and that all of the others are different. I’m just saying it’s evidence. We don’t have a lot of behavioral data on small animals anyways, but I think we can agree there’s probably a correlation between brain size and cognitive capacity.
***Do two pounds of “normal-sized” ants actually fit in a five-gallon bucket? Yes. I couldn’t find a number for “ant-packing density” in the literature, but thanks to the valiant efforts of David Manheim and Rio Lumapas, it seems to be between 0.3 gallons (5 cups) and 5.5 gallons. It depends on size and whether ants pack more like spheres or more like blocks.
Here’s a pattern I’d like to be able to talk about. It might be known under a certain name somewhere, but if it is, I don’t know it. I call it a Spaghetti Tower. It shows up in large complex systems that are built haphazardly.
Someone or something builds the first Part A.
Later, someone wants to put a second Part B on top of Part A, either out of convenience (a common function, just somewhere to put it) or as a refinement to Part A.
Now, suppose you want to tweak Part A. If you do that, you might break Part B, since it interacts with bits of Part A. So you might instead build Part C on top of the previous ones.
And by the time your system looks like this, it’s much harder to tell what changes you can make to an earlier part without crashing some component, so you’re basically relegated to throwing another part on top of the pile.
I call these spaghetti towers for two reasons: One, because they tend to quickly take on circuitous knotty tangled structures, like what programmers call “spaghetti code”. (Part of the problem with spaghetti code is that it can lead to spaghetti towers.)
Especially since they’re usually interwoven in multiple dimensions, and thus look more like this:
“Can you just straighten out the yellow one without touching any of the others? Thanks.”
Second, because shortsightedness in the design process is a crucial part of spaghetti machines. In order to design a spaghetti system, you throw spaghetti against a wall and see if it sticks. Then, when you want to add another part, you throw more spaghetti until it sticks to that spaghetti. And later, you throw more spaghetti. So it goes. And if you decide that you want to tweak the bottom layer to make it a little more useful – which you might want to do because, say, it was built out of spaghetti – without damaging the next layers of gummy partially-dried spaghetti, well then, good luck.
Note that all systems have load-bearing, structural pieces. This does not make them spaghetti towers. The distinction about spaghetti towers is that they have a lot of shoddily-built structural components that are completely unintentional. A bridge has major load-bearing components – they’re pretty obvious, strong, elegant, and efficiently support the rest of the structure. A spaghetti tower is more like this.
The motto of the spaghetti tower is “Sure, it works fine, as long as you never run lukewarm water through it and turn off the washing machine during thunderstorms.” || Image from the always-delightful r/DiWHY.
Where do spaghetti towers appear?
Basically all of biology works like this. Absolutely all of evolution is made by throwing spaghetti against walls and seeing what sticks. (More accurately, throwing nucleic acid against harsh reality and seeing what successfully makes more nucleic acid.) We are 3.5 billion years of hacks in fragile trench coats.
Scott Star Codex describes the phenomenon in neurotransmitters, but it’s true for all of molecular biology:
You know those stories about clueless old people who get to their Gmail account by typing “Google” into Bing, clicking on Google in the Bing search results, typing “Gmail” into Google, and then clicking on Gmail in the Google search results?
I am reading about serotonin transmission now, and everything in the human brain works on this principle. If your brain needs to downregulate a neurotransmitter, it’ll start by upregulating a completely different neurotransmitter, which upregulates the first neurotransmitter, which hits autoreceptors that downregulate the first neurotransmitter, which then cancel the upregulation, and eventually the neurotransmitter gets downregulated.
Meanwhile, my patients are all like “How come this drug that was supposed to cure my depression is giving me vision problems?” and at least on some level the answer is “how come when Bing is down your grandfather can’t access Gmail?
My programming friends tell me that spaghetti towers are near-universal in the codebases of large companies. Where it would theoretically be nice if every function was neatly ordered, but actually, the thing you’re working on has three different dependencies, two of which are unmaintained and were abandoned when the guy who built them went to work at Google, and you can never be 100% certain that your code tweak won’t crash the site.
I think this also explains some of why bureaucracies look and act the way they do, and are so hard to change.
I think there are probably a lot of examples of spaghetti towers, and they probably have big ramifications for things like, for instance, what systems evolution can and can’t build.
I want to do a much deeper and more thoughtful analysis about what exactly the implications here are, but this has been kicking around my brain for long enough and all I want to do is get the concept out there.
Does this feel like a meaningful concept? Where do you see spaghetti towers?



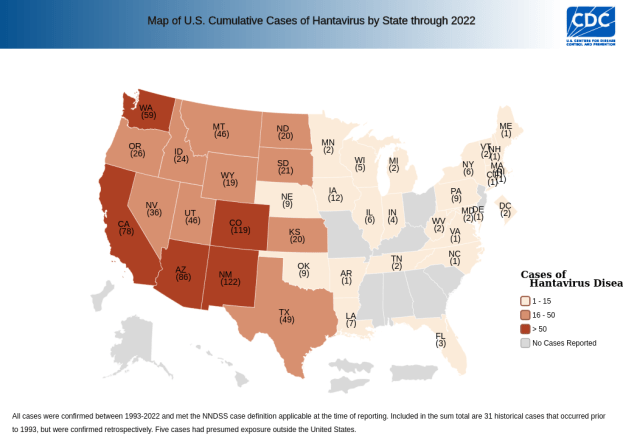





















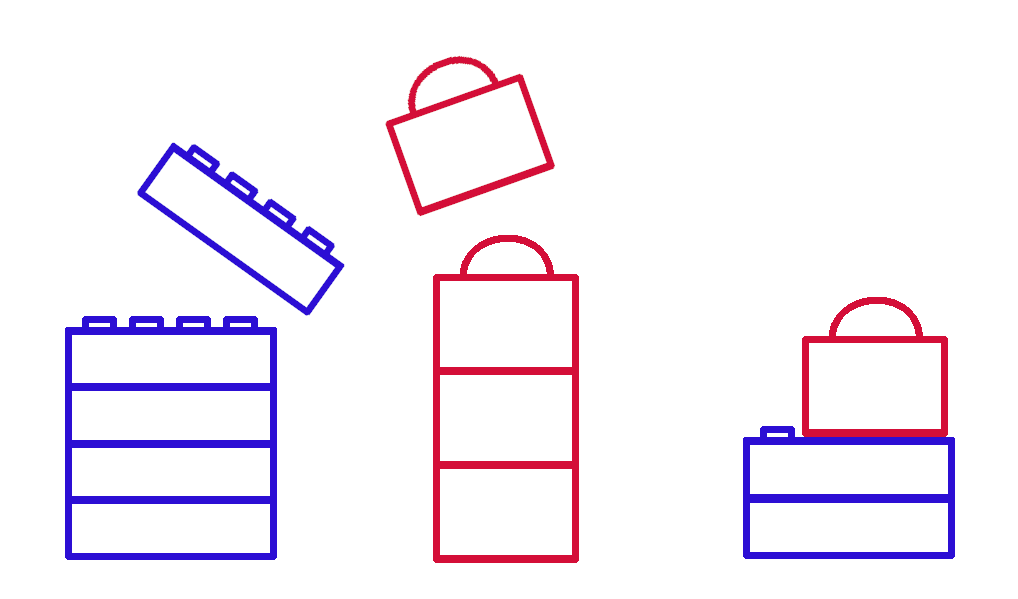












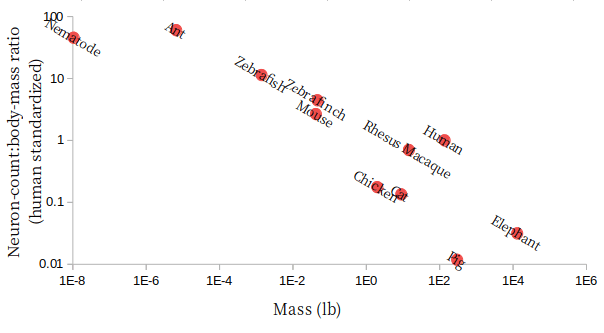




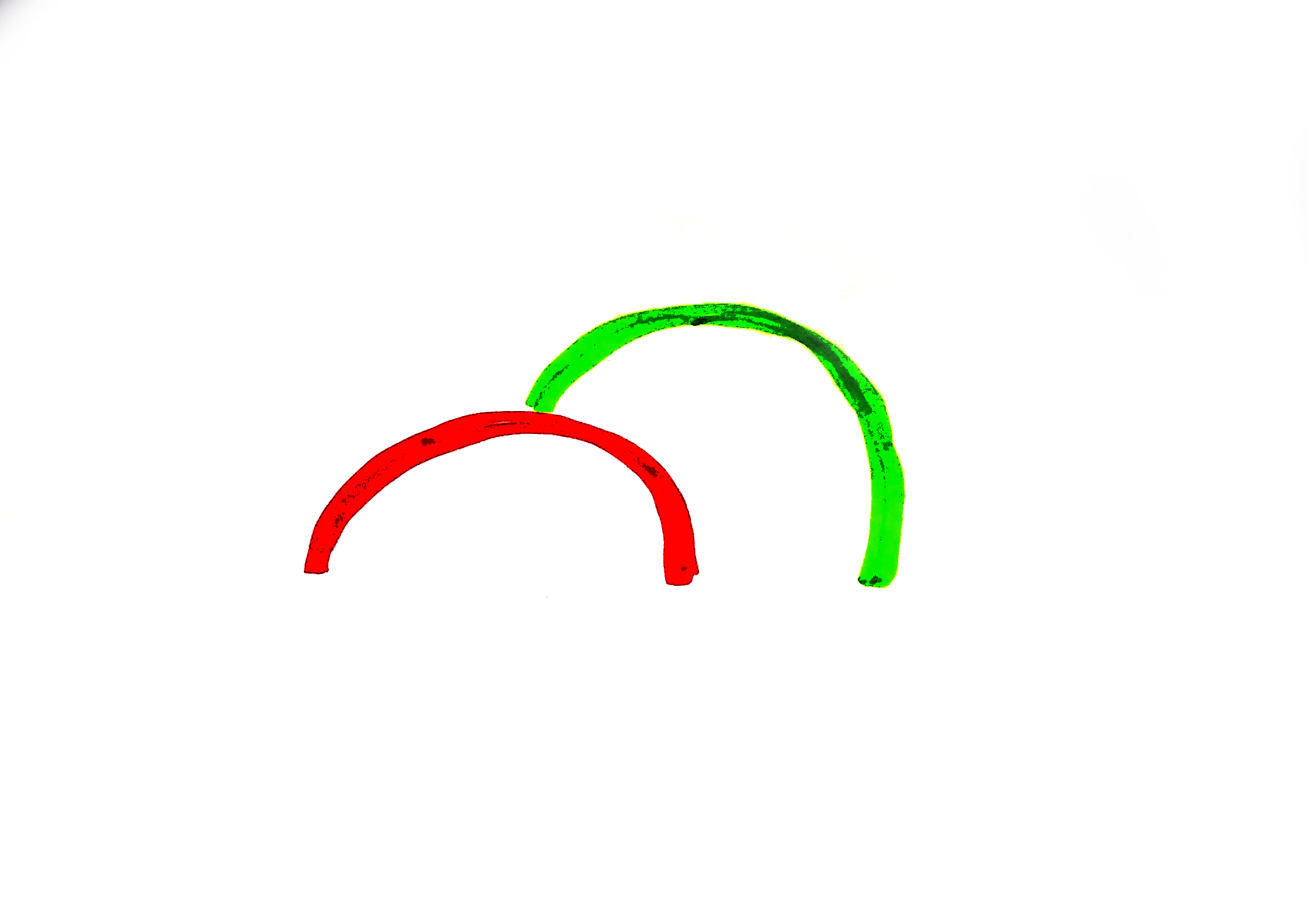





{kind=link}